
Abstract
The expression of a biologically active human IFNλ4 depends on the presence of a frameshift deletion polymorphism within the first exon of the interferon lambda 4 (IFNL4) gene. In this report, we use the lung carcinoma-derived cell line, A549, which is genetically viable to express a functional IFNλ4, to address transcriptional requirements of the IFNL4 gene. We show that the GC-rich DNA-binding transcription factor (TF) specificity protein 1 (Sp1) is recruited to the IFNL4 promoter and has a role in induction of gene expression upon stimulation with viral RNA mimic poly(I:C). By using RNAi and overexpression strategies, we also show key roles in IFNL4 gene expression for the virus-inducible TFs, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), IFN regulatory factor 3 (IRF3), and IRF7. Interestingly, we also observe that overexpression of IFNλ4 influences IFNL4 promoter activity, which may further be dependent on the retinoic acid-inducible gene-I (RIG-I)-like receptor pathway. Together, our work for the first time reports on the functional characterization of the human IFNL4 promoter.
Introduction
T
IFNL4 was discovered in the year 2013 by Prokunina-Olsson et al. during the course of their efforts to identify causal single-nucleotide polymorphisms (SNPs) at the IFNL locus that could be playing functional roles (25). IFNL4 was seen to be expressed as a protein of 179 amino acids (p179), in response to the viral RNA mimic poly(I:C), from primary hepatocytes derived only from those subjects carrying a deletion (ΔG) at the SNP site, rs368234815 (25). In addition to transcripts corresponding to p179, several different IFNL4 transcript isoforms were expressed in hepatocytes in response to poly(I:C) from both the ΔG allele and the alternate allele at the SNP site (TT) (25). However, the translated proteins from these other transcripts did not have homology with any known proteins and/or they had premature stop codons (25).
The 179 amino acid full-length IFNλ4 and the SNP giving rise to it (rs368234815) have since attracted attention from not only HCV researchers but also those involved with other viruses such as cytomegalovirus (CMV) (19). IFNλ4 (p179) has since been known to have antiviral activities against HCV and other flaviviruses (18), human corona viruses, and some human paramyxoviruses (1,8). While the role of IFNλ4 in the other viral infections is unclear, in HCV infections, it is known that expression of a functional IFNλ4 leads to a refractory state of IFN responsiveness in patients receiving IFNα-ribavirin combination therapy (3), thereby resulting in treatment failure. This has also been corroborated in in vitro studies where expression of an active IFNλ4 leads to stimulation of IFN-stimulated genes (ISGs) and renders the cells insensitive to IFNα (8,25).
Clearly, IFNλ4 seems to be having an important role in the innate immune responses raised by the host cells, especially those of the epithelial lineage against invading pathogens. Since, the expression of IFNλ4 is linked to expression levels of IFNλ3 due to linkage disequilibrium between rs368234815 and the two known putative causal SNPs, rs28416813 (4) and rs4803217 (20), the combined effect of IFNλ3 and IFNλ4 in the response to pathogens is likely to be complex. While much is needed to be studied on how the four IFNλs collaborate with each other and with other IFNs in responding to invading pathogens, there is no report yet on transcriptional regulation of the IFNL4 gene expression.
Studies on promoter characterization are routinely performed in established cell lines both with promoter–reporter constructs and to analyze transcriptional requirements in the genomic context. Since the expression of the full-length (p179) IFNλ4 depends on the presence of a deletion in its first exon, to study IFNL4 gene expression in the genomic context, one needs to first ascertain which cell lines have the genetic capacity for its expression and therefore are suitable to study its gene regulation and function. A recent report has shown that the lung carcinoma-derived cell line, A549, was able to express IFNL4 mRNA in response to human metapneumovirus (hMPV) infection (1). In addition, another recent report, which tested different cell lines, also saw that IFNL4 transcripts were generated only from A549 cells in response to poly(I:C) (a mimic of viral RNA) stimulation, suggesting that this cell line could be used to study the transcriptional regulation of the IFNL4 gene (18). Therefore, in this report, we used the A549 cell line to examine the different transcription factors (TFs) that could be functioning at the IFNL4 promoter to drive its expression. We also used promoter-driven luciferase constructs to further delineate the contours of the IFNL4 promoter using HEK293T cells.
Materials and Methods
Cell lines, plasmids, antibodies, and reagents
HEK293T and A549 cells were originally purchased from ATCC and maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum, 1× glutamax, 1× penicillin/streptomycin, and 1× fungizone, all of them procured from Invitrogen. Huh7.5 cells and the pJFH1 plasmid, from which HCV 3′ untranslated region (UTR) RNA was transcribed, were a generous gift from Charles Rice (The Rockefeller University, NY). pUNO and the genes encoding TFs, specificity protein 1 (Sp1), p50, p65, IFN regulatory factor 3 (IRF3), and IRF7 cloned in pUNO vector, were purchased from Invivogen. The constitutively active IRF constructs (IRF3-SA and IRF7-SA) and the pIFNβ promoter construct were generous gifts from Siddharth Balachandran (Fox Chase Cancer Center, PA). The gene encoding CEBP-α cloned in pCMV6.5 was from Origene Tech. IFNL4 cDNA was synthesized by GeneScript with codon optimization for expression in a baculovirus system. The plasmids—pMAVS, HCV RdRp clones, myeloid differentiation primary response gene 88 (MyD88), and RIG-I in pUNO vector—were kind gifts from Cheng Kao and Ranjith Kumar (Indiana University). Two different transfection reagents for both RNA (Lipofectamine 2000 from Invitrogen and TransMessenger from Qiagen) and DNA (Lipofectamine LTX-PLUS; Invitrogen and SuperFect, Qiagen) were used in the experiments. Poly(I:C) (low molecular weight) was obtained from Invivogen. Bacterial lipopolysaccharides (LPS) and betulinic acid (BA) were from Sigma. Mithramycin A (MA) was from Enzo Life Sciences. Power SYBR Green PCR Master Mix was from Applied Biosystems. The Ampliscribe RNA transcription kit (with T7 RNA polymerase) used to transcribe HCV 3′UTR from pJFH1 plasmid was from Epicentre. The chromatin immunoprecipitation (ChIP) kit was from Active Motif (ChIP-IT Express Enzymatic kit). All siRNAs were from SantaCruz Biotech. The antibodies against Sp1 (ChIP-certified rabbit polyclonal) were from Abcam. Antibody against hemagglutinin (HA) tag was from SantaCruz Biotech. 5′-ppp-dsRNA was purchased from Invivogen. Western blot chemiluminescence development kit was from Perkin Elmer. Dual-Glow Luciferase kit was from Promega. The oligonucleotide primers were synthesized from IDT Tech. All the enzymes used for DNA cloning were either from NEB or Invitrogen. All clones were sequenced by the Sanger dideoxy method as confirmation. pIFNL3 construct is the same as p1.4IL28B described in Chinnaswamy et al. (4). pIFNL4-1kb was constructed by cloning a 1,058 bp fragment of DNA amplified from genomic DNA isolated from human blood as described previously (4). The primers used for cloning this fragment and the deletion fragments are listed below (name of constructs as mentioned in Fig. 4D):
Construct A: Rev-5′-GATATCCTCGAGCTCTGCTTCTGCAGCAGGC-3′, For-5′-GATATCACGCGTTCGTGTGGCCTTTCGGG-3′;
Construct B: Rev-5′-GATATCCTCGAGCTCTGCTTCTGCAGCAGGC-3′, For-5′-GATATCGAGCTCAGGCGTTAGAGACC-3′;
Construct C: Rev-5′-GATATCCTCGAGCTCTGCTTCTGCAGCAGGC-3′, For-5′-GATATCGAGCTCGGGCAAAGCAAGACTCTG-3′;
Construct D: Rev-5′-GATATCCTCGAGCTCTGCTTCTGCAGCAGGC-3′, For-5′-GATATCGAGCTCGAAGATAAAGACAACCAGGGTG-3′;
Construct E: Rev-5′-GATATCCTCGAGCTCTGCTTCTGCAGCAGGC-3′, For-5′GATATCGAGCTCAAGCTTGCCCTGGACGGGAAAGCCCGGCTGCAAACCCCATTCTCAGC-3′; and
Construct F: Rev-5′-GATATCCTCGAGCTCTGCTTCTGCAGCAGGC-3′, For-5′-GATATCGAGCTCCCCATTCTCAGCCCTGCGC-3′.
The primers used to amplify DNA for in vitro transcription of HCV 3′UTR are For-5′-GATATCGAATTCGATTAATACGACTCACTATAGGGAGCGGCACACACTAGGTACAC-3′, Rev-5′-GATATCGGATCCACATGATCTGCAGAGAGACCAGTTAC-3′.
The primers used to test the mRNA levels of p50, p65, IRF3, IRF7, and Sp1 were derived from previous published studies and the sequences are listed in Supplementary Table S1 (Supplementary Data are available online at
In silico analysis of the IFNL4 promoter
The program Alibaba2.1 was run from the server:
Genotyping of A549 cell line
A549 cells were grown to 70–80% confluency in six-well plates. Genomic DNA was isolated from the cells using the QIAmp DNA blood midi kit (Qiagen). The genomic DNA was used to genotype rs368234815 using a competitive allele-specific PCR (KASP; LGC Genomics) assay using the manufacturer's instructions. The assay uses two fluorescent dyes, whose emissions were read by the StepOnePlus Real-Time PCR system (Applied Biosystems).
Semiquantitative reverse transcription-PCR
The RNA from A549 cells was purified using the RNeasy kit (Qiagen) and the purified RNA was converted to cDNA using the QuantiTect reverse transcription kit (Qiagen) that allows for effective elimination of gDNA contamination from RNA. The kit used random hexamer primers for the first-strand cDNA synthesis, and then gene-specific primers were used for PCR amplification of specific genes. The number of PCR cycles was standardized such that the amplification phase of different genes (IFNL4, IFNβ, IL15, and GAPDH) was stopped well before they reached the plateau stage so that comparisons between PCR amplicons from different RNA preparations could be made. Some reactions (as indicated in the figure legends) used the one-step RT-PCR kit (Qiagen), which uses gene-specific primer(s) for both cDNA synthesis and PCR amplification. Standardization of number of cycles was done as above for a semiquantitative comparison of PCR products arising from different preparations of RNA. The primer sequences (For-5′-TGAGGGATTTGGTAGTGAAAGG-3′; Rev-5′-TTAAACCGGGAGACAGAATAGC-3′) used for amplifying IFNL4 transcripts were kindly provided by A. Guerrero-Plata, Louisiana State University. The primers for other genes were as follows:
IFNL3: For-5′-CAGCTGCAGGTGAGGGAGCGCCCCG-3′,
Rev-5′-GGTGGCCTCCAGAACCTT-3′;
IFN-beta: For-5′-GATTCATCTAGCACTGGCTGG-3′,
Rev-5′-CTTCAGGTAATGCAGAATCC-3′;
GAPDH: For-5′-ATCCCATCACCATCTTCCAG-3′,
Rev-5′-CCATCACGCCACAGTTTCC-3′; and
IL15: For-5′-CATGGAGCACAGAAATCAATG-3′,
Rev-5′-TGGCTATGGCAAGGGGT-3′.
Real-time PCR (quantitative PCR)
For relative quantification of gene expression, quantitative PCR (qPCR) was done in the 7900HT fast real-time PCR system (Applied Biosystems) using Power SYBR Green PCR Master Mix (Applied Biosystems). The cDNA was prepared using the QuantiTect reverse transcription kit (Qiagen) that allows for effective removal of gDNA from RNA. For all reactions, a normal EtBr-gel-based PCR was tested to confirm that the primers amplified single bands and there were no primer dimers or any other nonspecific products before carrying out qPCR. GAPDH was used as housekeeping control. Triplicates were used for each reaction and the relative fold expression was calculated by the ΔΔCt method where ΔCt = CtDESIREDGENE − CtGAPDH. The primer sequences used to measure IFNL4 transcripts are shown under the Semiquantitative reverse transcription (RT)-PCR section above; the primers for measuring GAPDH transcript levels are For-5′-GGGTGTGAACCATGAGAAGTA-3′, Rev-5′-GGTGCAGGAGGCATTGCT-3′.
Chromatin immunoprecipitation
The ChIP assays were carried out from A549 cells grown in 15-cm plates stimulated with 25 μg/mL of poly(I:C) for 0, 3, 6, and 8 h, after which they were fixed in formaldehyde. After this stage, the assays were performed with the ChIP-IT express enzymatic kit (Active Motif) following the manufacturer's instructions. Three micrograms of the respective antibodies were used per chromatin preparation for immunoprecipitation. After reverse cross-linking and protease digestion, the DNA (both input and ChIP) was purified using the Active Motif's chromatin-IP DNA purification kit. To know the concentration of ChIP DNA, a standard curve was drawn. For this, qPCR was carried out in the 7900HT fast real-time PCR system (Applied Biosystems) using Power SYBR Green PCR Master Mix (Applied Biosystems). After obtaining the concentration of input DNA by the NanoDrop 8000 sectrophotometer (ThermoFischer), Ct values of serially diluted input DNA for each time point were used to draw the standard curves. The concentration of ChIP DNA was obtained by extrapolating their Ct values in the standard curves thus obtained. Standard curves were plotted at each time point for the input DNA by doing a serial dilution and the quantity of the ChIP DNA for Sp1 and IgG control was thus calculated and plotted as % of the input DNA. The primer sequences amplified the region −264 to −8 with respect to transcription start site (TSS) in the IFNL4 promoter. The primer sequences are For-5′-GATATCGAGCTCGAAGATAAAGACAACCAGGGTG-3′, Rev-5′-GACGGTCCATGCTGGCTTTTA-3′.
Western blot
HEK293T cells (4 × 105 cells/well) were seeded in six-well plates and grown overnight in complete DMEM at 37°C. At subconfluency, cells were transfected with 2 μg of plasmid/well containing the HA-tagged IFNL4 gene in pcDNA3.1 vector using SuperFect transfection reagent (Qiagen). Three hours later, media were changed. After 12 h of incubation, cells were washed with phosphate-buffered saline and lysed using 200 μL of RIPA buffer supplemented with protease inhibitor (Roche). The lysate was incubated at 4°C with shaking to ensure complete lysis (1 h). After incubation, lysate was centrifuged at 15,000 rpm for 10 min at 4°C in a refrigerated centrifuge (model No. 5424 R; Eppendorf). The supernatant was collected and protein estimation was done with bicinchoninic acid assay reagent (NEB). Thirty micrograms of whole cell lysate was loaded on 10% sodium dodecyl sulfate–polyacrylamide gel and transferred to a membrane (Immobilon PVDF membrane; Millipore) after electrophoresis along with a standard protein marker ladder. Western blotting was performed using antibody against HA tag (Santa Cruz Biotech), followed by incubation with secondary antibody (goat anti-mouse; Jackson Laboratory). The blot was developed using an enhanced chemiluminescence reagent kit (PerkinElmer) and visualized by ChemiDoc (Bio-Rad).
Electrophoretic mobility shift assay
Nuclear extract preparation
Subconfluent HEK293T cells grown in 100-mm dishes were transfected with 5 μg/dish of pCMV-p65 plasmid for 24 h. Transfected cells were then harvested and lysed with 500 μL of Buffer A (10 mM HEPES, pH 8.0, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, and 0.5% NP40) and nuclei were pelleted by centrifugation at 10,000 rpm for 20 min. Nuclear pellets obtained in this manner were then incubated with 500 μL of Buffer B (20 mM HEPES, 25% glycerol, pH 8.0, 0.42 mN NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 0.5 mM phenylmethanesulfonyl fluoride (PMSF); PIC). Nuclear fractions obtained by centrifugation at 10,000 rpm for 30 min were dialyzed against Buffer C (0.2 mM EDTA, 20% glycerol, 20 mM HEPES, pH 8.0, 0.1 M KCl, 1 mM DTT, and 1 mM PMSF) overnight, centrifuged for a further 10,000 rpm for 30 min, and used in experiments. Target oligos from IFNL4 promoter, 5′AAGCTTGCCCTGGAC
Luciferase assays
The luciferase assays were done in COSTAR (Corning, Inc.) 96-well plates (white, clear bottom) as described previously (4), unless otherwise specified (for some assays, cells were grown in six-well plates, as indicated in the figure legends). Briefly, 20–25 × 103 cells/well were seeded and grown for 24 h before carrying out transfections. Transfections were carried out with Lipofectamine LTX and PLUS Reagent (Invitrogen) according to the manufacturer's instructions. Typical reactions included 10 ng/well of firefly luciferase reporter construct and 0.01 ng/well of a control plasmid expressing Renilla luciferase (pRLTK). Luciferase assays were carried out 24 h post-transfection using the Dual-Glo Luciferase assay kit (Promega) on a GLOMAX 96-well plate reader with dual-injection system (Promega). Firefly luciferase readouts were normalized to Renilla luciferase values (plotted as FF/Re. Luc. Activity) before the analysis.
Results
Structural and functional characterization of the IFNL4 promoter
We used in silico prediction methods to identify putative TF binding sites in the proximal promoter of IFNL4 (Fig. 1A). We focused on the promoter and UTR DNA about 1 kb upstream of the start codon of IFNL4. We chose this length of DNA based on the observation in a previous report, which showed that 1 kb promoter and UTR DNA upstream of the start codons of IFNL1, 2, and 3 were sufficient for maximal transcription efficiency in reporter constructs (13). The computer prediction that allowed for a minimum matrix conservation of 75% (see the Materials and Methods section for other parameters used) revealed the presence of several TF binding sites (Supplementary Table S2) within the ∼1 kb promoter and UTR DNA of IFNL4. A CATA box (CATAAAA) instead of the conventional TATA box (TATAAAA) is present at the IFNL4 promoter, 25 bp upstream of the TSS (Fig. 1A, boxed sequence). No other TATA-like element is observed in the immediate vicinity around the TSS (within 30 bp upstream and downstream). Of the different TFs predicted to bind to the promoter, we were interested in the virus-induced TFs and therefore we concentrated on Sp1, NF-κB, and IRF binding sites (interferon-stimulated response element [ISREs]). There were several Sp1 [21] and a few NF-κB [3] and IRF binding sites [4] that were predicted (Fig. 1A). Along with these four TFs, we also considered CEBP-α for further analysis as it had the maximum number [15] of predicted binding sites of all the different TF binding sites predicted apart from Sp1 (Supplementary Table S2).

Structural and functional characterization of IFNL4 promoter.
We cloned a DNA fragment of 1,058 bp situated upstream of the start codon of IFNL4 that contained 274 bp DNA from 5′UTR and 784 bp DNA from the promoter region into pGL3basic vector upstream to the firefly luciferase gene. This reporter construct, referred to as pIFNL4-1kb, and its derivatives were used in the rest of the study in in vitro luciferase assays.
The RIG-I (retinoic acid-inducible gene-I)-like receptor (RLR) pathway senses viral replication intermediates in the cytosol and triggers a cascade of events involving the adaptor protein, mitochondrial antiviral signaling protein (MAVS). The viral RNA-dependent RNA polymerases (RdRps) can generate 5′ triphosphorylated small-RNA molecules that can efficiently stimulate RIG-I to signal through MAVS, leading to activation of NF-κB and IRFs (26). A reporter construct carrying a promoter with the binding sites for TFs that are activated by the RLR pathway (mainly NF-κB and IRFs) can be used to assay the promoter activity in cells transiently transfected with the RdRps (4,26). This system that used the IFNβ promoter–firefly luciferase gene construct as the reporter has been well-tested for the RdRps from HCV, norovirus, classical swine fever virus, and brome mosaic virus (16,26,29,30,31). In fact, this system utilizing the HCV RdRp also could stimulate the IFNL3 promoter activity (4). To test if the IFNL4 promoter construct (pIFNL4-1kb) could functionally respond to signaling by the HCV RdRp through the RLR pathway, we overexpressed RdRps from different genotypes of HCV (genotype 1a, 2a, and 3a) in the presence of an exogenously provided RIG-I, in HEK293T cells. The pIFNL4-1kb construct was transfected along with a transfection control (Renilla luciferase) and luciferase activity was measured in the cells 24 h post-transfection (Fig. 1B). We saw that of the three RdRps tested, the RdRp from HCV genotype 3a showed the maximal activity of the IFNL4 promoter, while the one from genotype 2a had intermediate activity, and genotype 1a RdRp did not show any significant activity emanating from the IFNL4 promoter. While these differences could arise from the amount of RIG-I ligands the three RdRps were able to generate, importantly, this assay shows that the IFNL4 promoter could respond to RIG-I signaling similar to the IFNβ and IFNL3 promoters (4,26).
Huh7.5 cells are liver cancer-derived cells, which support replication of HCV due to a mutation in the RIG-I gene that debilitates the RLR pathway (32). We next tested the pIFNL4-1kb construct in these cells to know if it can respond to synthetic 5′-ppp-dsRNA (a ligand for RIG-I) delivered into the cells by transfection (Fig. 1C). As expected, no induction of promoter activity was seen after transfecting 5′-ppp-dsRNA alone. However, when the ligand was combined with an overexpressed WT RIG-I gene, we saw an induction in activity of the IFNL4 promoter.
These results that utilized two different sources of RIG-I ligands (HCV RdRp and exogenously provided 5′-ppp-dsRNA) in two different cell lines (HEK293T and Huh7.5) suggested that the RLR pathway can trigger the IFNL4 promoter and an intact RLR pathway can generate signals to activate the proximal promoter of IFNL4 in transient transfection assays.
The genotypic status of rs368234815 in A549 cells and its utility as a tractable model to study IFNL4 expression
While the RLR pathway functions in the cytosol, the toll-like receptor (TLR) pathway comprising the TLR3 and TLR4 molecules can sense viral RNA and bacterial cell wall components, respectively, from outside the cells and can lead to transcription activation of IFN and other cytokine genes. Since it was recently reported that the lung carcinoma-derived A549 cell line could express IFNλ4 (1,18), we used this cell line to study IFNL4 expression. First, we stimulated A549 cells with poly(I:C) (a viral RNA mimic) and LPS (bacterial-derived lipopolysaccharide and a ligand for TLR4) and analyzed the expression of IFNL4 mRNA by RT-PCR (Fig. 2A). We used the primer set designed by A. Guerrero-Plata and colleagues—for their study on hMPV infection of A549 cells that give rise to a PCR product of 120 bp after binding to the 3′UTR of the IFNL4 mRNA (1)—in our RT-PCR. This primer set binds to several of the transcripts generated from expression of the IFNL4 gene, including the transcripts encoding for p179 arising from the ΔG allele at rs368234815 (Table 1). We treated the cells with poly(I:C) and LPS for 12 h in the media and measured the IFNL4 transcript levels in a semiquantitative way using the GAPDH gene expression as the standard. There was an induction of IFNL4 expression only in the treated cells, while the control cells did not show such expression (Fig. 2A).
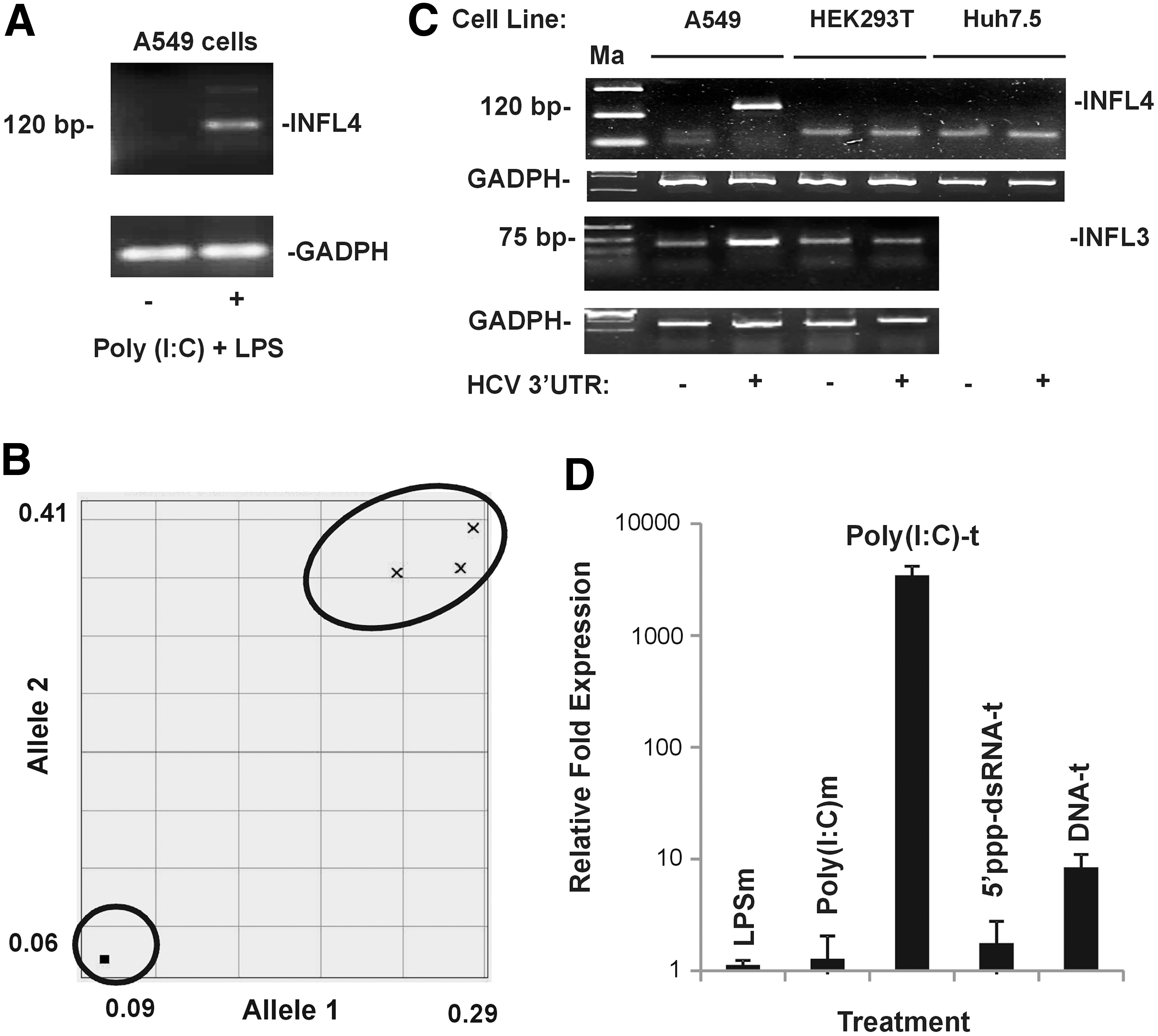
A549 cells are ideal to study IFNL4 expression.
The names of the corresponding proteins translated from the respective transcripts are shown. Of all the proteins derived from the transcripts, only p179 is known to be functional (shown in bold).
UTR, untranslated region.
Since IFNλ4 is expressed as a result of a frameshift arising due to a deletion (ΔG) at the SNP site, rs368234815 (25), we were interested to know the status of this SNP in the A549 cell line. For this purpose, we performed a competitive allele-specific PCR assay (KASP; LGC Genomics) on the genomic DNA isolated from A549 cells (Fig. 2B). From this assay, it was clear that the A549 cell line was heterozygous at rs368234815 (Fig. 2B). The above results show that the A549 cells contain at least one copy of the ΔG allele that may be giving rise to the RT-PCR product seen in Figure 2A. It is possible that along with transcripts of p179, transcripts of other nonfunctional IFNλ4 isoforms (Table 1) may also be expressed in A549 cells upon stimulation with viral RNA/RNA mimics. While a study dealing with the alternate pre-mRNA splicing mechanisms leading to production of different IFNL4 isoforms under different stimulation conditions is beyond the scope of the current study, nevertheless, the A549 cell line gives us an opportunity to study the induction mechanism of the IFNL4 promoter.
We next used two other cell lines, Huh7.5 (human hepatocarcinoma derived) and HEK293T (human embryonic kidney origin), along with A549 cells to study IFNL4 expression. We used the HCV 3′UTR RNA, a ligand for RIG-I (28), as the viral RNA mimic for this experiment. An in vitro transcribed (by T7 polymerase, Ampliscribe; Epicentre) HCV 3′UTR RNA was transfected into the three cell lines, and after 12 h, the cells were lysed and examined for IFNL4 expression by semiquantitative RT-PCR. It was clear from this experiment that only A549 cells, but not Huh7.5 and HEK293T, were able to give rise to the 120 bp specific product representing IFNL4 transcripts (Fig. 2C, top). The failure of Huh7.5 and HEK293T cells to express IFNL4 could be because these cells may lack the adequate signaling mechanism by the RIG-I pathway. While it is known that Huh7.5 cells are deficient in RIG-I signaling (32) and therefore they may not express IFNL4 upon HCV 3′UTR stimulus, no information is available in this regard for HEK293T cells. We therefore assessed if HEK293T cells were also deficient in expression of another IFNL, IFNL3, compared with A549 cells. Our results indicated that HEK293T cells did not express endogenous IFNL3 in response to transfected HCV 3′UTR, while a clear stimulation was evident for the A549 cell line (Fig. 2C, bottom). The above results convinced us that the A549 cell line is ideal to study innate immune signaling leading to expression of IFNL genes, particularly IFNL4.
We next used different means of activating the IFNL4 transcription in A549 cells. We treated the media with LPS (LPSm); poly(I:C) [poly(I:C)m]; transfected poly(I:C) [poly(I:C)-t]; transfected 5′-ppp-dsRNA (5′-ppp-dsRNA-t); and transfected empty pUNO plasmid DNA (DNA-t) and measured IFNL4 expression by qPCR (Fig. 2D). We saw that transfecting poly(I:C) showed the strongest induction of all the methods used and surprisingly naked plasmid DNA also showed substantial amount of IFNL4 expression.
Sp1 is recruited to the IFNL4 promoter upon poly(I:C) stimulation
We overexpressed six TFs, CEBP-α, Sp1, p50 (NF-κB1), p65 (REL-A), IRF3, and IRF7 in A549 cells, and stimulated them with poly(I:C)m and LPSm and measured IFNL4 expression by semiquantitative RT-PCR (Fig. 3A). We used the empty pUNO vector as control. None of the overexpressed TFs, except the GC-rich DNA-binding TF Sp1, were able to significantly increase the expression of IFNL4 over that of the vector control in either the presence or absence of poly(I:C) and LPS. Sp1 showed an increase in IFNL4 expression over the vector control only in the presence of poly(I:C)m and LPSm. To confirm this stimulation, we overexpressed Sp1 at increasing concentrations in A549 cells both in the presence and absence of poly(I:C)m and LPSm (Fig. 3B). We were able to see a dose-dependent effect of Sp1 on IFNL4 expression only in the presence of poly(I:C)m and LPSm. This suggested that Sp1 gets activated by the stimulus provided, likely by some post-translational modifications, and is able to increase transcription from the IFNL4 promoter. To see if this effect was specific for IFNL4 gene expression, we measured the levels of the IFNβ expression under the same conditions of increasing concentration of overexpressed Sp1 (Fig. 3C). No induction of the IFNβ gene was observed in these conditions (missing band shown by the arrow at the 186 bp position specific for the IFNβ gene transcript, Fig. 3C). However, an Sp1-dependent cytokine IL15 (5) was induced by the overexpressed Sp1 (Supplementary Fig. S1).
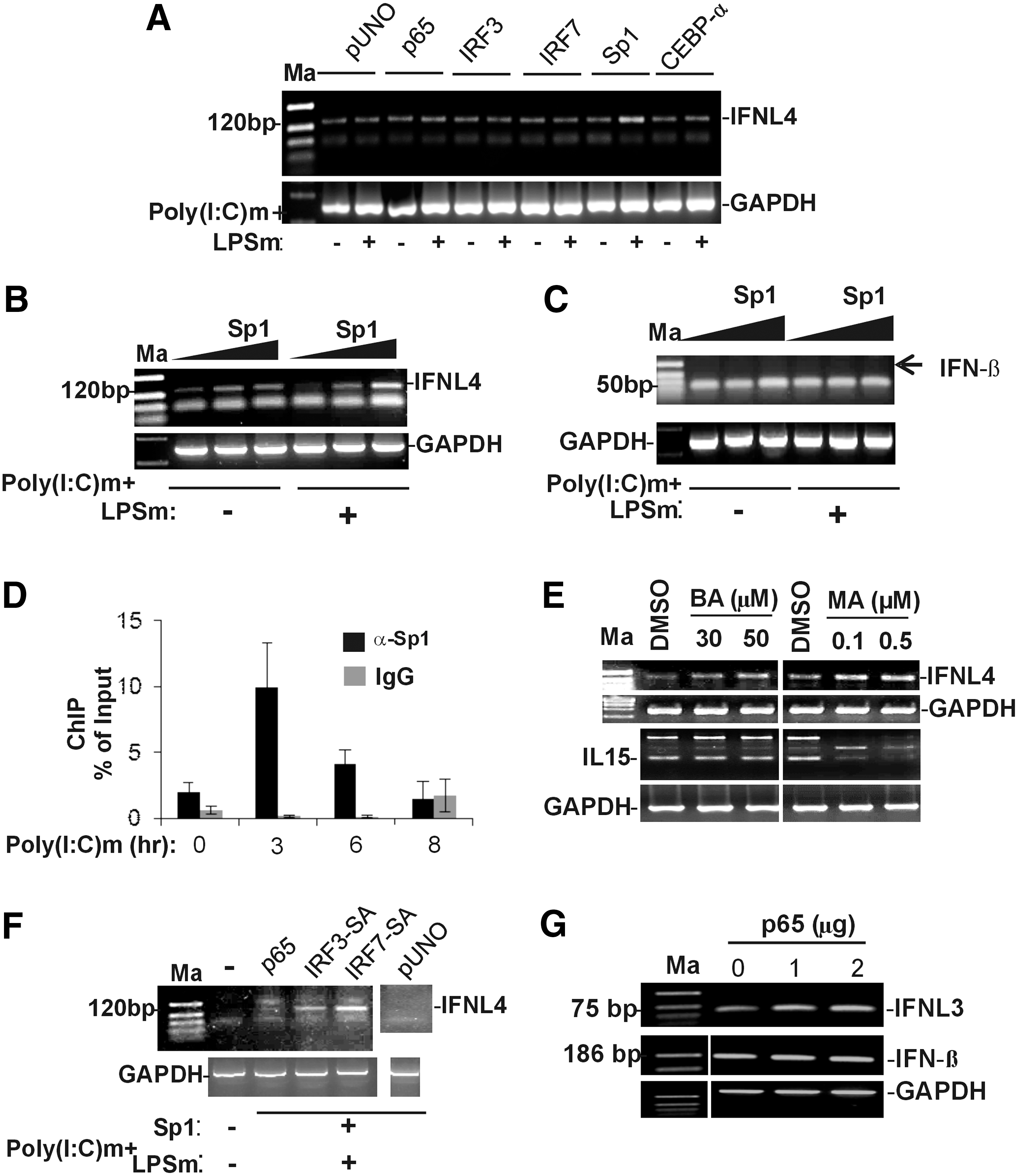
Sp1 is recruited to the IFNL4 promoter and induces its transcription.
To confirm the role of Sp1 at the IFNL4 promoter, we carried out ChIP assays. We stimulated A549 cells with poly(I:C)m for 0, 3, 6, and 8-h intervals and harvested cells before performing ChIP assays using a polyclonal antibody against Sp1. The influenza virus HA-specific IgG was used as the control antibody. qPCR assays were performed with the immunoprecipitated DNA by using primers binding to a region around the TSS. Figure 3D shows that Sp1 is recruited to the IFNL4 proximal promoter upon poly(I:C) stimulation. Maximal occupancy is seen within 3 h of stimulation, while it declines at 6–8 h poststimulation (Fig. 3D). This result clearly shows the role of Sp1 at the IFNL4 promoter upon stimulation by poly(I:C)m, explaining the results seen in Figure 3A and B.
To further understand the significance of Sp1 in IFNL4 expression, we used Sp1-specific inhibitors, betulinic acid (BA) and mithramycin A (MA), which are also known anticancer drugs. BA leads to ubiquitination and proteasome-mediated degradation of Sp1, and MA binds to GC-rich DNA, preventing binding of Sp1 to its binding sites on DNA (10,34). We first treated A549 cells with the two inhibitors at two concentrations, and then after changing media, we stimulated the cells with poly(I:C)m. The cells were harvested after 12 h and IFNL4 expression was assayed by semiquantitative RT-PCR (Fig. 3E). The results showed that BA instead of inhibiting IFNL4 expression increased it at both the concentrations tested. Similarly, MA also increased IFNL4 expression. We also assayed for mRNA levels of IL15, an Sp1-dependent cytokine (5), in similar conditions and saw that only MA was able to inhibit IL15 expression, but not BA. These results suggest that either there is a TF redundancy at the IFNL4 promoter or that the nonspecific effects of the two drugs caused an increase in IFNL4 expression and that the regulation of the IFNL4 promoter could be complex. Interestingly, a recent report showed that IFNλ4 has antiproliferative properties (24), which seem to agree with our results where MA, being an anticancer drug, increases IFNL4 gene expression. Similarly, another report (17) showed that BA has anti-HCV property, and it is possible that BA induces IFNλ4 for this function. Nevertheless, more specific Sp1 inhibitors have to be tested to understand the effect of inhibition of Sp1 on the IFNL4 gene expression.
The failure by the other TFs tested (CEBP-α, p50, p65, IRF3, and IRF7) to increase IFNL4 expression was surprising. While CEBP-α could be a TF that is used during developmental stages and may not be a virus-induced TF that can be activated by poly(I:C), it was expected that p65 (p50 is not always a positive regulator of transcription) IRF3 and IRF7 should be able to exert an effect on IFNL4 expression. This is based on what has been observed for the expression of the other three IFNLs, IFNL1, 2, and 3, under conditions of poly(I:C)m stimulation carried out in a hepatocyte-derived large T antigen transformed cell line, PH5CH8 (13). Our results suggested that either these TFs are inadequately activated by the stimuli provided or that their binding sites are not available for occupancy at the IFNL4 promoter. Previous studies suggest that poly(I:C)m cannot activate TFs required for IFNβ expression in several hepatocarcinoma-derived cell lines (14). To overcome this potential problem in A549 cells, we transfected plasmids coding for constitutively active IRF3 and IRF7 (IRF-3SA and IRF-7SA) into the A549 cells before stimulating the cells with poly(I:C)m and LPSm. Under these conditions, we saw that both the constitutively active IRFs were able to increase IFNL4 expression substantially (Fig. 3F). p65, on the other hand, again failed to show any IFNL4 expression. Since no specific post-translational modifications are needed for p65 to translocate to the nucleus to activate target genes except for the requirement that it has to be separated from the IκB complex, our results suggest that overexpressed p65 may not be able to bind to its potential target in the IFNL4 promoter. This is in contrast to its effect on the IFNL3 transcription, where a dose-dependent increase in IFNL3 transcripts was observed by overexpressed p65 (Fig. 3G), although a similar increase in IFNβ transcripts was not observed. Together, the above results showed that activated Sp1, IRF3, and IRF7 can stimulate IFNL4 expression in A549 cells. On the other hand, overexpressed p65 is not sufficient to increase IFNL4 expression in A549 cells, unlike its effect on IFNL3 expression.
Sp1, NF-κB, IRF3, and IRF7 are required for IFNL4 expression
To unambiguously know the requirement of each of the TFs, we next carried out siRNA-mediated knockdown of these TF genes, and then assayed for IFNL4 expression under activating conditions using semiquantitative RT-PCR. For stimulating IFNL4 expression, we transfected poly(I:C) into A549 cells as this treatment gave maximal induction of IFNL4 expression compared with other methods (Fig. 2D). We confirmed that each of the five TFs was knocked down sufficiently by siRNA treatment so as to see an effect on IFNL4 expression (Fig. 4B). From this experiment, it was clear that p50, p65, Sp1, IRF3, and IRF7 knockdown adversely affected IFNL4 expression in A549 cells in response to poly(I:C)-t (Fig. 4A). Surprisingly, knockdown of none of the above TFs negatively impacted the expression of IFNβ. We observed in in vitro promoter assays that the IFNβ promoter is far more active than both IFNL3 and IFNL4 promoters (Supplementary Fig. S2B), which may be a reflection of likely affinities for the respective promoters for different target TFs in the RLR pathway. Since our siRNA knockdown efficiency was not 100% (Fig. 4B), we speculate that the residual TFs are sufficient to transcribe from the IFNβ promoter, but much higher concentrations are needed for optimal IFNL4 gene transcription. Nonetheless, the results clearly showed roles for Sp1, NF-κB, IRF3, and IRF7 in transcription of the IFNL4 gene in response to poly(I:C).
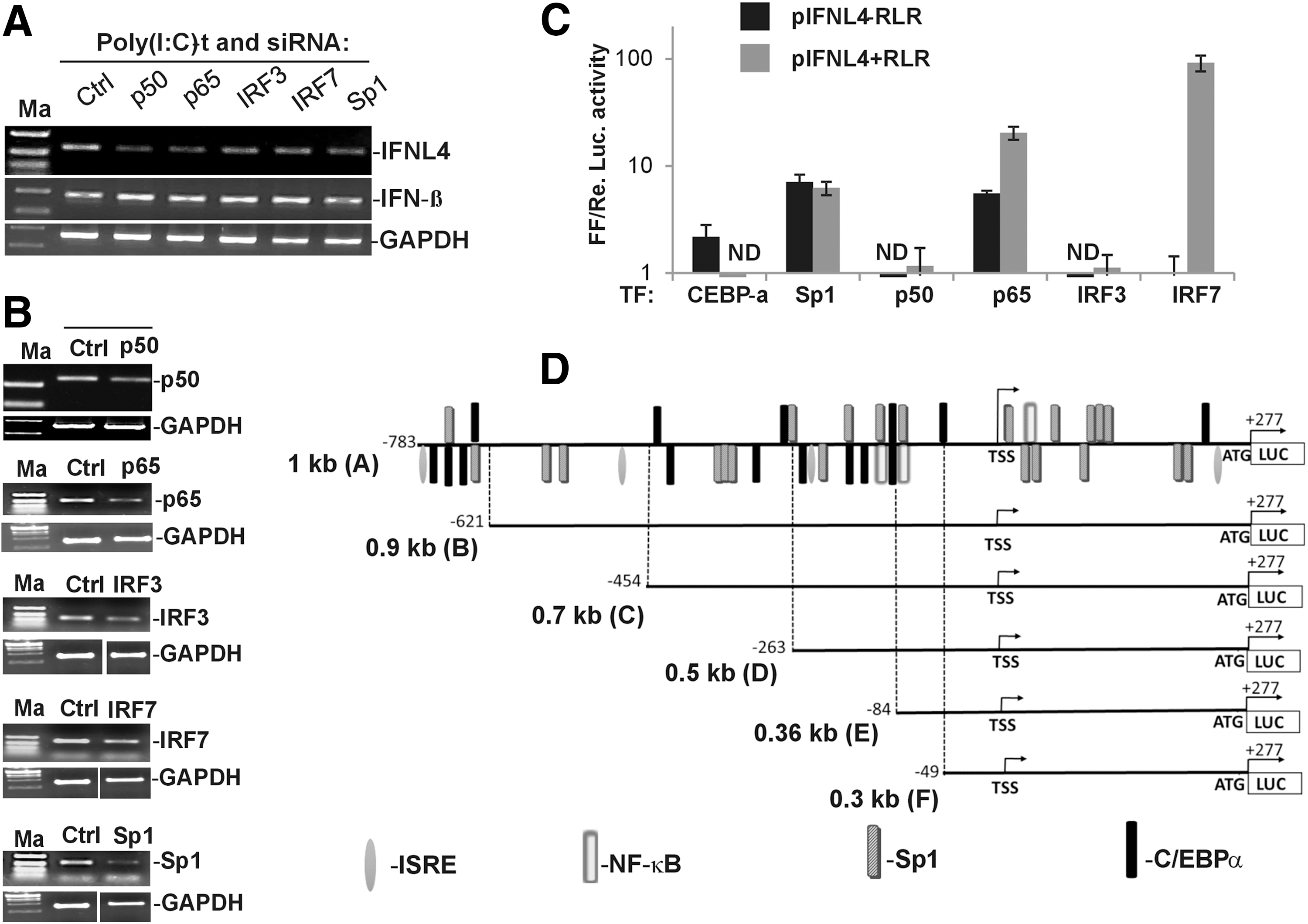
Sp1, NF-κB1, IRF3, and IRF7 are required for IFNL4 expression.
We next used the construct pIFNL4-1kb that was described in Figure 1B to examine the effect of overexpression of each of these TFs on the promoter activity. Compared with the results that we obtained from A549 cells so far on IFNL4 expression from the genomic context, the reporter construct is a tool to examine the effect of individual TFs on the promoter activity in activating and nonactivating conditions in a nongenomic context. We used the HCV RdRp to activate the RLR pathway and provide activating signals for the overexpressed TFs. All the six TFs (CEBP-α, Sp1, p50, p65, IRF3, and IRF7) were overexpressed in the presence or absence of a co-overexpressed RIG-I and HCV RdRp (+ and −RLR) and assayed for luciferase activity (Fig. 4C). The results showed that both CEBP-α and Sp1 could individually stimulate the IFNL4 promoter activity, which was not enhanced by activation signals provided by the HCV RdRp and RIG-I. p50 did not influence the promoter activity in both activating and nonactivating conditions, while p65 showed a stimulation of IFNL4 promoter activity in both conditions. The strongest influence from activating the RLR pathway was on the effect of IRF7 on the promoter activity of IFNL4. IRF7, in the presence of HCV RdRp and exogenously provided RIG-I, showed more than a 90-fold increase in stimulation of the IFNL4 promoter activity. IRF3 did not show any significant effect on the IFNL4 promoter activity in the luciferase assays compared with IRF7.
Deletion analysis of the IFNL4 promoter
To delineate the contours of the IFNL4 promoter in its responses to these TFs, we made several deletion constructs of pIFNL4-1kb (Fig. 4D). Each of these deletion mutants harbored a different number of the predicted TF binding sites. First, we used the pIFNL4-1kb and its derivative deletion constructs to assess the effect of CEBP-α on the IFNL4 promoter activity in HEK293T cells under nonactivating conditions (-RLR) (Fig. 5A, bottom left). The 1 kb construct has 15 CEBP-α binding sites, which gradually decrease toward the 3′ end, and the shortest construct (construct F, Figs. 4D and 5A) has a single predicted binding site for CEBP-α. In agreement with this prediction, the deletion constructs showed a decreasing level of IFNL4 promoter activity as the 5′ end got truncated and it was minimal with construct F (Fig. 5A), suggesting that there is an additive effect on the promoter activity from the different binding sites over the length of the promoter. The Sp1 TF was predicted to have 22 binding sites distributed evenly along the length of the 1kb construct (construct A, Figs. 4D and 5A). The deletion constructs were transfected along with Sp1 and no RLR pathway activation was provided. The constructs showed a pattern of IFNL4 promoter activity, which suggested that even the shortest construct (construct F) tested had activity comparable with the longest one (construct A) (Fig. 5A, bottom right). If we presume that no additive effect on the promoter activity may be present between the individual Sp1 binding sites, then this result is in agreement with the prediction of Sp1 binding sites as construct F still had ∼45% of the predicted binding sites. Since equal concentrations of the different constructs were used and the DNA concentration was not normalized to the length of the constructs used in the assays, the pattern seen in the luciferase activities of the different constructs in Figure 5A, bottom right, is expected. Whether an additive effect is lacking between the individual Sp1 binding sites cannot be demonstrated by this assay and needs a different set of experiments to ascertain this.
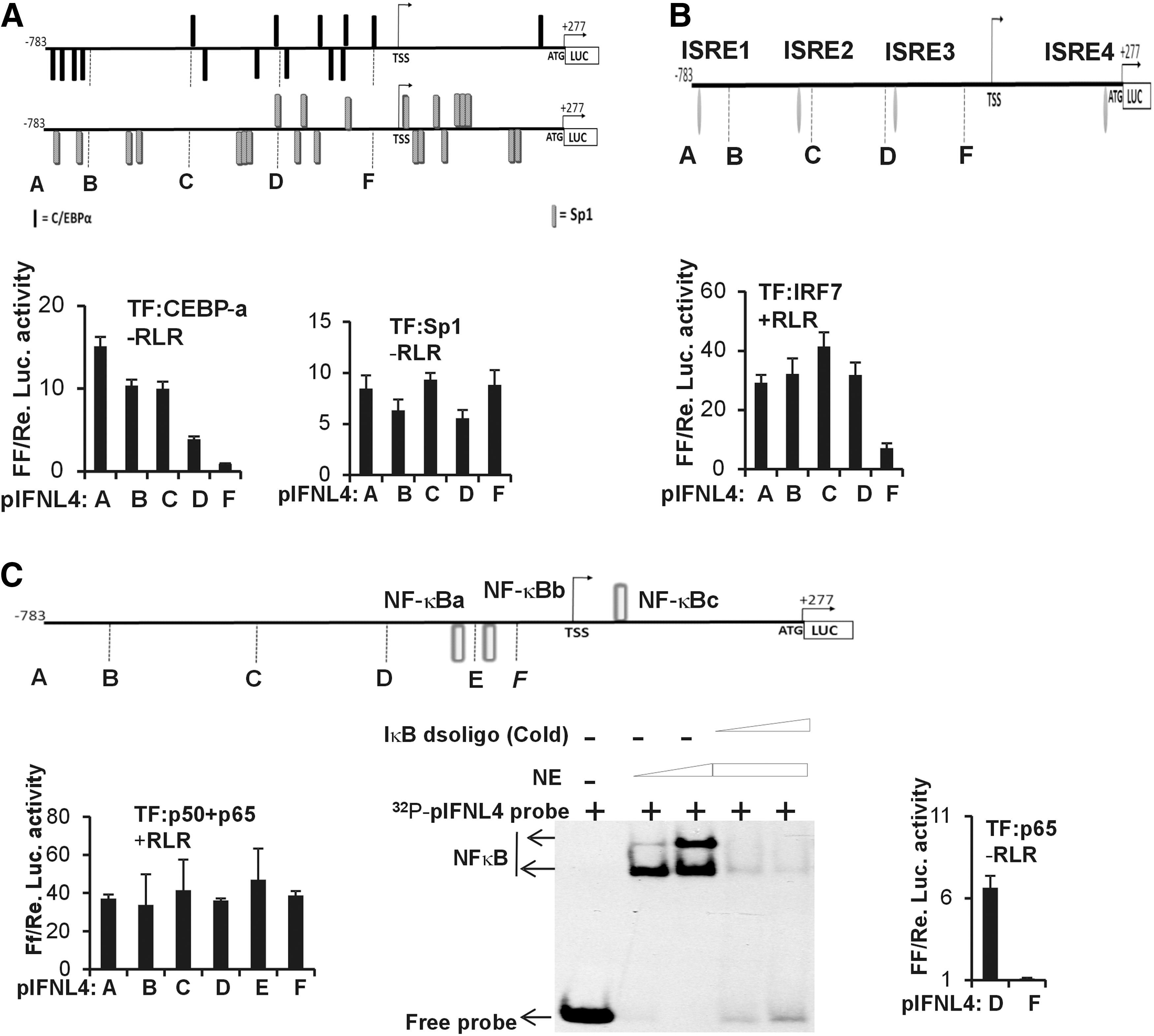
Deletion analysis of the IFNL4 promoter.
We next tested the deletion constructs against overexpressed IRF7. Since IRF7 showed the maximal effect due to an active RLR pathway (Fig. 4C), we provided activating conditions by overexpressing HCV RdRp and RIG-I. Under these conditions, it was evident that maximal IFNL4 promoter activity was achieved by deletions up until construct D (Figs. 4D and 5B, bottom). Construct D included two of the four predicted IRF binding sites (ISRE1–4) (Fig. 5B, bottom). Construct F, which had only one of the four predicted IRF binding sites (ISRE4), was not sufficient for maximal promoter activity.
Next, we tested for the effect of overexpression of NF-κB against promoter activity in the different deletion constructs. Since p50 and p65 combination is the relevant form of NF-κB in viral infections, we co-overexpressed equal amounts of the two gene constructs so that p50-p65 heterodimers are formed and translocate to the nucleus to activate the IFNL4 promoter. The RLR pathway was also activated by HCV RdRp and RIG-I. The results showed a pattern similar to what was observed with Sp1 (compare Fig. 5C bottom left and 5A bottom right), with the shortest construct (construct F) showing as much activity as the longest one (construct A), suggesting that there is no additive effect on the promoter activity from the different binding sites over the length of the promoter, unlike the effect seen with CEBP-α (Fig. 5A). As per the predictions, construct F had only one predicted NF-κB binding site (NF-κBc). From this assay, it is difficult to differentiate whether the observed pattern of promoter activity with the deletion constructs is due to lack of an additive effect between the individual TF binding sites or it is merely because some of the predicted binding sites lack efficient TF binding capacity. It is possible that the other two sites (NF-κBa and b) may not be contributing to NF-κB binding and promoter activity and that only NF-κBc is functional. To test this possibility, we performed an electrophoretic mobility shift assay (EMSA) using nuclear lysates of HEK293T cells where p65 was overexpressed. We used a radiolabeled probe comprising the region of DNA that included NF-κBb binding site (Fig. 5C, middle). Our results showed that NF-κBb binding site could bind to p65 under in vitro conditions. Interestingly, this sequence (of NF-κBb binding site) was previously identified in a rat gene promoter as capable of binding NF-κB (22). Furthermore, we saw that this NF-κB binding site has high degree of conservation with a site in the proximal promoter region of the porcine IFNL4 gene, which was also predicted to bind NF-κB (Supplementary Fig. S3). The porcine IFNL4 gene is not a pseudogene, unlike its status in the human population. The conservation of this site across the two species suggests that it may be functionally relevant. To further confirm the roles of the two NF-κB binding sites (NF-κBa and b), we transfected constructs D and F in HEK293T cells and overexpressed p65 alone without p50. No activating conditions were provided. We saw that the deletion of NF-κBa and NF-κBb severely crippled the ability of the promoter to respond to overexpressed p65 (Fig. 5C, right). Since we saw that co-overexpression of p50 and p65 has a different effect on the promoter than overexpressing p65 alone (Fig. 5C, bottom left and bottom right), we do not completely understand this result at this point unless we perform additional experiments. This may likely be due to different binding affinities of the three binding sites for p65 homodimers versus p50-p65 heterodimers.
Further mutational analysis with the reporter constructs will reveal the roles of the individual TF binding sites in driving transcription from the IFNL4 promoter.
IFNλ4 stimulates its own promoter through the RLR pathway
A recent report had shown that overexpressing IFNλ4 had an inhibitory effect on the IFNL3 promoter activity (21). We were interested to test the effect of an overexpressed IFNλ4 on the IFNL4 promoter activity. For this purpose, we cloned the full-length IFNL4 cDNA sequence in pCDNA3.1 vector and tagged it with the coding sequence for an HA tag at the 3′ end. Since no reliable antibodies are yet available against the IFNλ4 protein, we confirmed the expression of IFNλ4 from our construct in HEK293T cells by probing for the HA tag, which is expressed at the C-terminus of IFNλ4 (Fig. 6A).
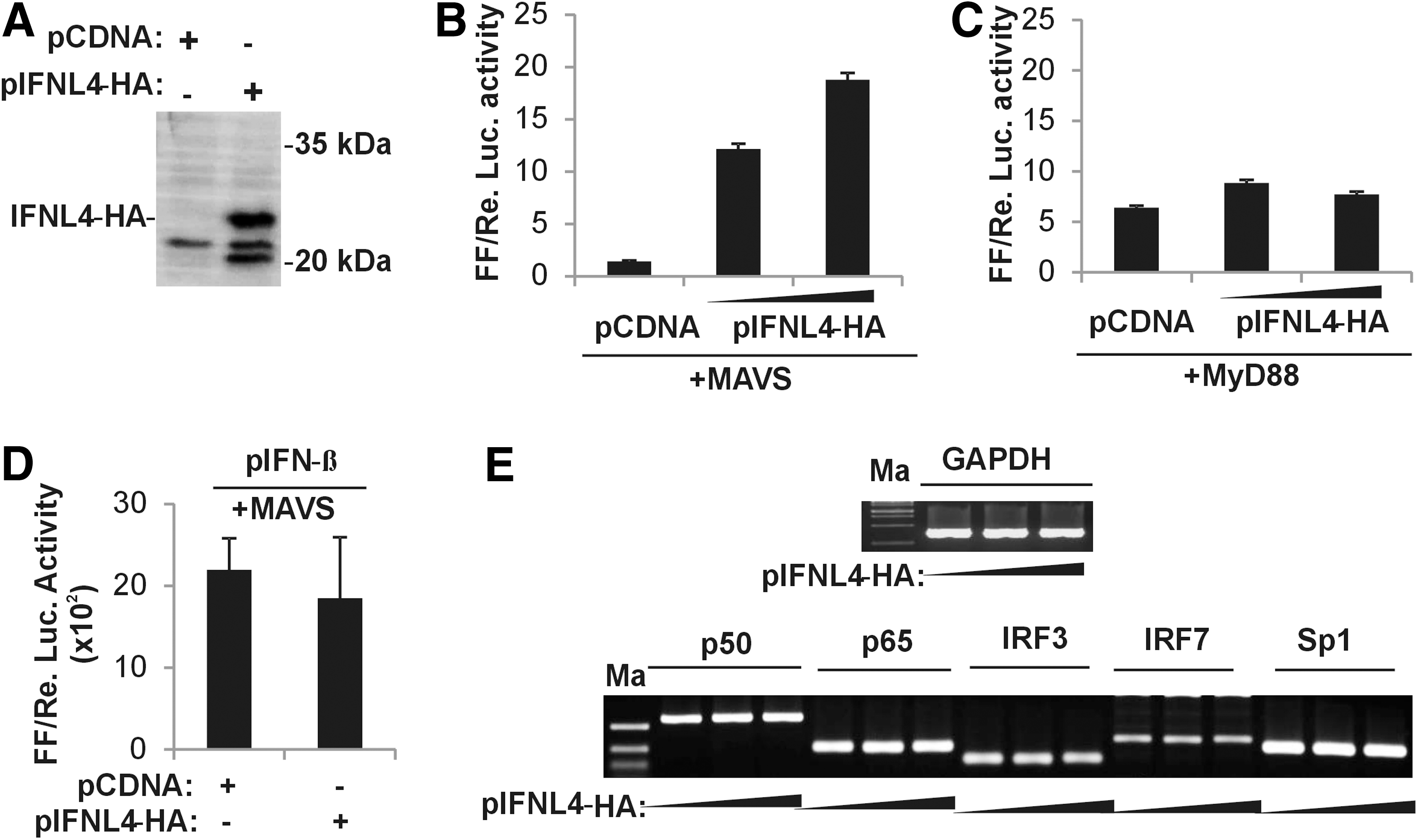
IFNλ4 stimulates IFNL4 promoter activity through the RLR pathway.
Based on our findings so far, the two predominant signaling pathways operating within the cytoplasm that may lead to induction of the IFNL4 promoter may be the RLR and the TLR pathways. MAVS is an adaptor protein in the RLR pathway, while TRIF (TIR domain-containing adapter-inducing IFNβ) and MyD88 are adaptor proteins in the TLR pathway. The TLR pathway also leads to activation of the TFs, NF-κB and IRF3/7, similar to the RLR pathway. To test the effect of the IFNλ4 protein on the IFNL4 promoter activity, which may be mediated by the above two signaling pathways, we first overexpressed MAVS and supplemented it with IFNL4-HA at two different concentrations (Fig. 6B). Under these conditions, we assayed for the activity of the pIFNL4-1 kb construct. Luciferase assays carried out after 24 h post-transfection showed that overexpressed IFNL4-HA had a stimulatory effect on the IFNL4 promoter when MAVS was overexpressed (Fig. 6B). Excluding MAVS from the transfection mix prevented the same effect (Supplementary Fig. S2A). We next repeated the experiment by replacing MAVS with MyD88 (Fig. 6C). It was observed that IFNL4-HA failed to have a stimulatory effect on the IFNL4 promoter in the presence of the TLR pathway adaptor, MyD88 (Fig. 6C). These results further emphasize the importance of the RLR pathway in stimulating the IFNL4 promoter. However, IFNL4-HA failed to simulate the IFNβ promoter in the presence of overexpressed MAVS (Fig. 6D). If IFNL4-HA stimulated the RLR pathway, then the effect should also be reflected in the IFNβ promoter activity, which is also responsive to the RLR pathway. Although we do not fully understand this, we do note that the IFNβ promoter is a much stronger promoter compared with both IFNL3 and IFNL4 promoters under in vitro conditions (Supplementary Fig. S3B).Therefore, the quantitative effects (such as the binding affinities for the different TFs) of any stimulatory signals on each of the promoters could be very different, resulting in different levels of effects on the two promoters. Although our data do not fully explain how IFNλ4 could be stimulating the IFNL4 promoter, it is possible that it could increase the expression of the different TFs that are functioning at the IFNL4 promoter. This may be especially true for the TF IRF7, which is considered an ISG itself, and since IFNλ4 leads to induction of several ISGs, an increase in IRF7 expression may be expected. To test this possibility, we overexpressed IFNL4-HA in A549 cells without any other stimulus and measured the transcript levels of the five TFs that we identified to be functioning at the IFNL4 promoter (Fig. 6E). We saw that none of the TFs, including IRF7 expression, were affected by the overexpression of IFNλ4. This would suggest that IFNλ4 functions at a different level in the pathway, most likely at the MAVS level. It will be interesting to further dissect the mechanism of stimulation of IFNλ4 on the IFNL4 promoter activity, in which MAVS seems to be an integral player.
Discussion
Type III IFNs were discovered a little over a decade ago and have come to the forefront of IFN research in the last few years. GWAS in chronic HCV infections in the year 2009 implicated IFNλs as critical determinants of a successful host immune response to HCV (3). The IFNL4 gene was recently discovered and found to be expressed only in some individuals who carry a deletion that gives rise to a frameshift in the coding region, leading to the translation of IFNλ4 with 179 amino acids (25). The in vivo mechanism of action is not clear for this newly identified member of the IFNλ family (23). However, it is known to be responsible for expression of high levels of ISGs in patients with chronic HCV infections who do not respond to conventional therapy (3,23).
IFNλ4 is an intriguing IFN with atypical features such as its inefficient secretion from expressing cells and its detrimental role in HCV infections (8,18). Its secretion depends on its glycosylation status, while its activity does not (8). On the other hand, it has clear antiviral properties when expressed inside cells, while this property is a disadvantage in patients with chronic HCV infections (3,18). Initial speculations held that IFNλ4 may use only the IFNλ-R1 receptor and not use the IL10-R2 receptor, unlike other IFNλs where both receptors are used for antiviral signaling; later research has shown that IFNλ4 indeed uses both the receptors for its activity (8). These features of IFNλ4 have aroused interest in its role in the pathogenesis mechanisms in diseases involving various viral infections. The SNP rs368234815 present in the first exon of IFNL4, which is responsible for the generation of the full-length active IFNλ4, has been actively pursued in genetic association studies in both viral and nonviral infections (7,19,27).
The transcriptional regulation of IFNL1, 2, and 3 has been well studied (11,13). Specific binding sites for NF-κB, IRF3, and IRF7 have been identified in their proximal promoter regions (13). IFNL1 promoter is also known to have a cluster of distal enhancers comprising of NF-κB binding sites that are needed for full induction of the promoter (33). However, no report is yet available on the transcriptional regulation of IFNL4. Since IFNλ4 has about 41% amino acid similarity with IFNλ3 and has unambiguous antiviral properties against several viruses, it is expected that its transcriptional regulation may also be similar to other IFNλs (8,18,25). However, since expression of IFNλ4 is disadvantageous to suppress viral replication in patients with chronic HCV infections, some differences in TF requirements for IFNL4 promoter and the other IFNL promoters are expected given the functionally disparate roles for IFNλ4 and the other IFNLs in suppressing HCV replication (3). Furthermore, the IFNL4 gene, including its promoter region, is located on a CpG island in the distal promoter region of IFNL3 (2). Therefore, IFNL4 promoter requirements may not exactly be the same as that of the other IFNL promoters at least in terms of virus-induced TFs.
In this report, we show that apart from NF-κB, IRF3, and IRF7, the GC-rich DNA-binding TF, Sp1, has a key role in stimulating transcription from the IFNL4 promoter. We saw that Sp1 was recruited to the IFNL4 promoter maximally within 3 h of stimulation with poly(I:C) added in the media (Fig. 3D). Furthermore, overexpression of Sp1 leads to stimulation of IFNL4 expression in the presence of poly(I:C)m in A549 cells (Fig. 3A, B), while it leads to stimulation of the promoter in the reporter construct pIFNL4-1kb in HEK293T cells (Fig. 4C). Although we included LPSm along with poly(I:C)m in some of our experiments, poly(I:C)m was sufficient to recruit Sp1 to the IFNL4 promoter (Fig. 3D). Last, siRNA knockdown of Sp1 debilitated the expression of IFNL4 in response to intracellular delivery of poly(I:C) (Fig. 4A). These results show a novel role for Sp1 TF in the expression of IFNL4. Whether the remaining IFNLs, especially IFNL3 (since its distal promoter overlaps with IFNL4 proximal promoter), also use Sp1 for transcription is yet to be studied. Sp1-mediated transcription inhibitors, BA and MA, failed to inhibit IFNL4 expression in our assays (Fig. 3E). We do not understand this result clearly, but it is likely that the two drugs used are nonspecific in their effects and their indirect effects on the transcription of other genes (e.g., BA can stimulate transcription of NF-κB) (12) are affecting IFNL4 transcription. This also suggests that the regulation at the IFNL4 promoter may not be simple, but involves a complex interplay of multiple TFs.
Our results from different experiments show that the three virus-induced TFs that drive the transcription of the other three IFNLs—NF-κB, IRF3, and IRF7 (13)—also can drive transcription from the IFNL4 promoter. Even though we did not see any IFNL4 transcripts upon overexpression of p65 in A549 cells either in the presence or absence of poly(I:C)m (Fig. 3A), several alternate lines of evidence show a role for NF-κB in IFNL4 expression in different conditions. First, we show by EMSA that p65 can bind to a bonafide NF-κB binding element (NF-κBb) present in the proximal promoter of IFNL4 (Fig. 5C). We then show that reporter constructs lacking NF-κBa and b sites are severely debilitated for promoter activity under conditions of p65 overexpression (Fig. 5C). Complementing the result that overexpression of p65 can stimulate IFNL4 promoter in the pIFNL4-1kb construct (Fig. 4C), the siRNA knockdown experiment clearly indicates that both p50 and p65 are required for optimal IFNL4 transcription when poly(I:C) is transfected into the cells, which likely activates both the RLR and TLR3 (endosomal) pathways (Fig. 4A).
Similarly, our results clearly show that IRF3 and IRF7 can stimulate IFNL4 transcription in the genomic context in A549 cells (Fig. 3F). Furthermore, overexpressed IRF7 shows strong induction of the IFNL4 promoter activity only when the RLR pathway is activated (Fig. 4C), suggesting clearly that this pathway has a predominant role in regulating the IFNL4 promoter. Deletion analysis of the IFNL4 promoter shows that a ∼540 bp fragment of DNA upstream of the IFNλ4 start codon is required for maximal activity of the promoter induced by IRF7(Fig. 5B). The siRNA knockdown experiment also showed that both IRF3 and IRF7 are required for optimal expression of IFNL4 (Fig. 4A). Our results also show that the TF, CEBP-α, can stimulate transcription from the IFNL4 promoter; however, it is not affected by the RLR pathway (Figs. 5A and 4C). In summary, we show by using the A549 cell line, which has the genetic potential to express the full-length IFNλ4, the different TFs that participate in its transcription. Although the primer set used in the study can bind to all the transcripts generated by the two alleles (ΔG and TT) at the SNP rs368234815 and not solely to the transcript responsible for the functional IFNλ4 protein, our study reports on the different TFs that are responsible for transcription of the IFNL4 gene as a whole. This is a limitation of our study. Furthermore, it is unlikely that different transcription induction mechanisms would be functional in generating the different IFNL4 isoforms arising from the ΔG allele-carrying copy of the gene (as ΔG allele is present in the exon and not on the promoter); rather it may be the result of different splicing mechanisms in operation. This topic may be an interesting area of research in future. We complement our results from A549 cells with the reporter construct studies in the HEK293T cell line to further characterize the IFNL4 promoter.
An interesting observation in our report is the stimulation of the IFNL4 promoter activity by IFNλ4 protein. We do not fully understand the mechanism of this stimulation yet, but our results show that the adaptor protein, MAVS, plays a key role in this effect (Fig. 6B, C). It is possible that IFNλ4 interacts with the MAVS protein and affects its active conformation (MAVS is known to function as prion-like aggregates upon stimulation of the RLR pathway) (9), leading to stimulation of the IFNL4 promoter by activating the downstream RLR pathway. This will be an interesting area of future research to understand the complicated role of IFNλ4 protein in the pathogenesis mechanism involving chronic HCV infections.
Footnotes
Acknowledgments
The authors thank Prof. Partha P. Majumder, director, National Institute of Biomedical Genomics, Kalyani, for his support and encouragement and Siddharth Balachandran, Fox Chase Cancer Center, PA, for the critical reading of the manuscript and gift of IRF3-SA, IRF7-SA, and pIFNβ plasmids. The authors thank Gopal Chakrabarti, Dept. of Biotechnology, University of Calcutta, WB, India, for sharing the A549 cell line. The authors thank Prof. Marek Kowalski, chair, Dept. of Immunology, Rheumatology and Allergy, Medical University of Lodz, Poland, for his immense help with the KASP assay and also thank Joanna Rywaniak for help in arranging the figures. A part of this work was presented at the annual meeting of the International Cytokine and Interferon Society, Cytokines 2015, in Bamberg, Germany. The authors would like to acknowledge Elsevier for permitting the use of the conference abstract in this report. A.B. acknowledges the junior research fellowship awarded by the Dept. of Biotechnology, Govt. of India. The funding for this work was provided to S.C. by the Dept. of Biotechnology, Govt. of India, as grant No. BT/PR7035/MED/12/579/2012.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.