
Abstract
Development of cervical cancer is associated with persistent infections by high-risk human papillomavirus (HPV). Although current HPV L1-based prophylactic vaccines prevent infection, they do not help to eliminate prevalent infections or lesions. Our aims were (i) to generate a vaccine combining prophylactic and therapeutic properties by producing chimeric capsomers after fusion of the L1 protein to different fragments of E2 from HPV 16, and (ii) to evaluate their capacity to generate an antitumoral cellular response, while conserving L1 neutralizing epitopes. Chimeric proteins were produced in Escherichia coli and purified by glutathione S-transferase (GST)-affinity chromatography. Their structure was characterized using size exclusion chromatography, sucrose gradient centrifugation, electron microscopy, and anti-L1 enzyme-linked immunosorbent assay. All chimeric proteins form capsomers and heterogeneous aggregates. One, containing part of the carboxy-terminal domain of E2 and its hinge region (L1Δ+E2H/NC, aa 206–307), conserved the neutralizing epitope H16.V5. We then evaluated the capacity of this chimeric protein to induce a cytotoxic T-cell response against HPV 16 E2. In 51Cr release cytotoxicity assays, splenocytes from C57BL/6 immunized mice recognized and lysed TC-1/E2 cells, which express and present endogenously processed E2 peptides. Moreover, this E2-specific cytotoxic response inhibited the growth of tumors of TC-1/E2 cells in mice. Finally, we identified an epitope (aa 292–301) of E2 involved in this cytotoxic response. We conclude that the L1Δ+E2H/NC chimeric protein produced in bacteria can be an effective and economically interesting candidate for a combined prophylactic and therapeutic vaccine that could help eliminating HPV16-positive low-grade cervical lesions and persistent viral infections, thus preventing the development of lesions and, at the same time, the establishment of new infections.
Introduction
T
Virus-like particles (VLPs) and capsomers formed by the L1 protein from HPV 16 have been proposed for the delivery of viral tumor epitopes and as platform for prophylactic and therapeutic vaccines, since they can induce both humoral and cellular immune responses (18,22,34,35). Chimeric VLPs consisting of the viral E7 (aa 1–60) protein fused to the carboxyl terminus of the L1 protein have been shown to be safe and immunogenic in preclinical studies and a clinical phase I trial (24). Capsomers can be an advantageous alternative to VLPs, because of the low cost and relatively simple production in Escherichia coli, and the fact that they are very stable and conserve their neutralizing epitopes (6,41,58).
E2 is an HPV protein that plays important roles in the viral life cycle. In HPV 16, it is composed of three main regions denoted N-terminal, hinge, and C-terminal domains; the N-terminal domain is composed of N1 and N2 subdomains and a fulcrum motif.
The E2 protein is expressed in low-grade squamous intraepithelial lesions (LSIL) (13,32,57), and it has been shown to be involved in the immune response against papillomaviruses, as suggested in preclinical studies, where tumor regression was associated with a specific immune response against the E2 protein of the cottontail rabbit papillomavirus (CRPV) (44); and by regression of rabbits' papillomas induced by immunization with the CRPV E2 protein (45). In another model, immunization with an adenovirus-based vaccine expressing the canine oral papillomavirus E2 protein, conferred protection against wart formation in beagle dogs after virus challenge (23).
In humans, clinical studies have shown an HPV 16 E2-specific IFN-γ-secreting T-cell response in healthy females, in contrast to the poor response of women with HPV 16+ cervical cancer (10,11); and the development of delayed type hypersensitivity reaction when a pool of E2 peptides of HPV 16 was applied in healthy women, while women with cervical neoplasia did not (48).
Interestingly, patients who resolved cervical dysplasia had higher T-cell proliferative responses against HPV 16 E2 than those whose dysplasia persisted, indicating an association between E2-specific proliferative responses and disease regression (12). Another study in a large group of women with LSIL, monitored during 1 year, showed that in those who regressed the lesion displayed strong E2-specific T-cell response assessed by IFN-γ ELISPOT, while in patients who progressed to high grade squamous intraepithelial lesion (HSIL) did not (54).
Therefore, induction of an HPV 16 E2-specific cellular response could constitute an adjuvant in treatment for HPV lesions, as shown when patients with high grade vulvar intraepithelial lesions with E2-, E6-, and E7-specific T-cell responses, remit after Imiquimod treatment more often than those without such responses; this was also seen in women with HPV 16+ vulvar intraepithelial neoplasia, who presented viral clearance and anti-E2 T-cell responses, Th-1 type cytokine production (IFNγ/IL2), after surgical or local topical treatment; whereas no response was observed in women with persistent lesions (21).
All these data support use of the E2 protein from HPV 16 as a target for therapeutic vaccination to help cure persistent HR HPV infections and lesions derived therefrom. Here, we fused different fragments of the HPV 16 E2 protein to the C-terminal domain of a modified L1 to generate constructs expressing chimeric proteins that were produced in E. coli, purified and characterized. The protein containing L1 fused to the hinge region and a sequence from the carboxy-terminal domain of the E2 protein (E2 206–307aa, HPV 16 L1Δ+E2H/NC), retained a neutralizing epitope of L1. It was used as antigen for further analyses. We show that immunization with this chimeric protein induces cytotoxic T-cell responses against E2 expressing tumor cells and inhibits tumor growth in C57BL/6 mice. In addition, we identified an epitope of E2 (292–301aa) involved in this cytotoxic response.
Materials and Methods
Construction of bacterial expression vectors
Fragments from the HPV 16 E2 gene were cloned into pGex 4T-2 L1ΔN10ΔC29 plasmid [previously described in (42)], downstream of the glutathione S-transferase (GST) tag and the L1 genes, using restriction enzymes NsiI/HindIII. The L1 protein expressed from this vector lacks the first 10 and the last 29 aminoacids of the native L1 protein from HPV 16 (L1ΔN10ΔC29 called L1Δ from here). E2 gene was obtained from plasmid pBR322-HPV 16 (15). The chimeric constructs generated are depicted in Figure 1.
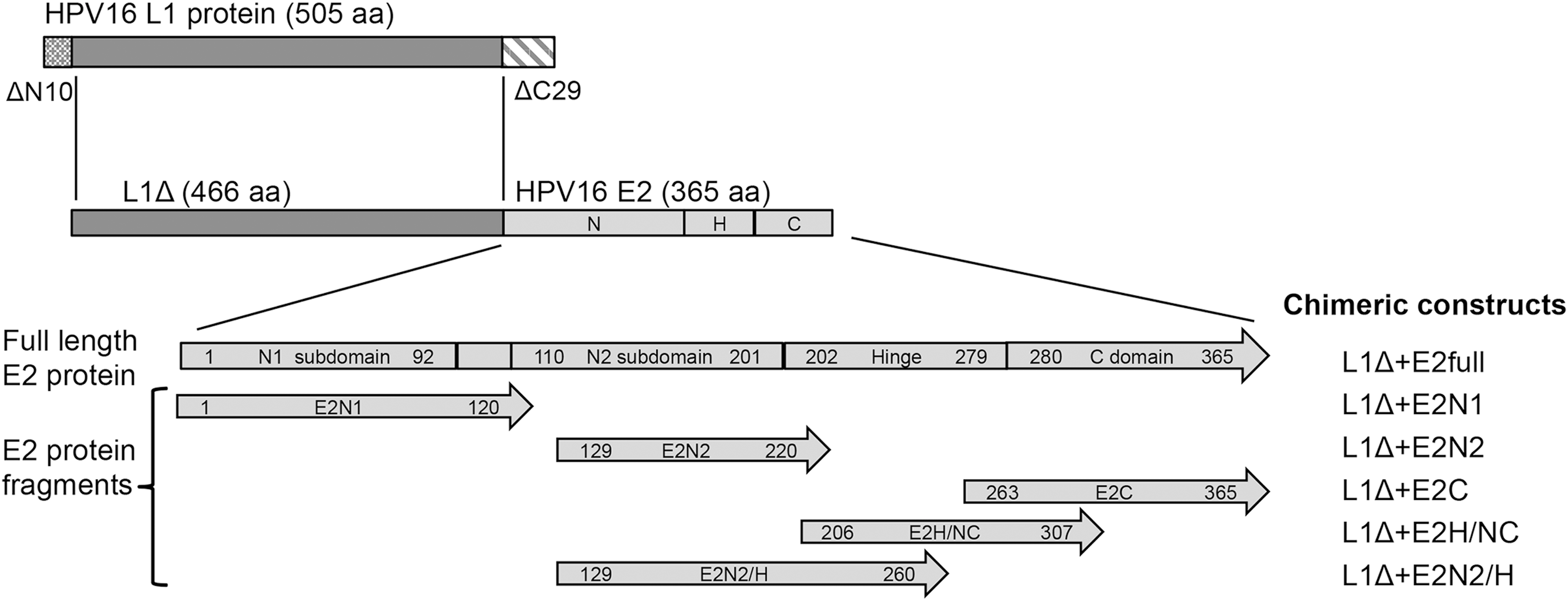
Chimeric constructs. A modified HPV 16 L1 protein (L1Δ) lacking the 10 amino-terminal aminoacids (ΔN10) and the 29 carboxy-terminal aminoacids (ΔC29) was fused with E2 full-length protein or five different E2 fragments. A full-length E2 protein depicting its native domains is shown. First and last aminoacid residues of E2 protein included in each construction are indicated within arrowed boxes; C, carboxy; H, hinge; HPV, human papillomavirus; N, amino.
Production and purification of L1Δ+E2 chimeric proteins
Proteins were produced as GST+L1Δ+E2 fusions, by a modified version of the method reported by Chen et al. (6). Briefly, E. coli Rossetta was transformed with the vectors mentioned above and grown at 37°C, 200 rpm in Terrific Broth medium supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Once the culture reached an optical density of 0.4 at 600 nm, isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.2 mM and the culture was further incubated overnight at 25°C and 200 rpm. Cells were harvested and the purification was carried out using 5-mL GSTrap (17513102; GE Healthcare) affinity chromatography columns. Eluates containing L1Δ+E2 chimeric proteins were loaded on a size exclusion chromatography column. Output fractions were analyzed by 12% denaturalizing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blot (WB) anti-HPV 16 L1 (CamVir-1; Pharmingen).
The fraction with a greater amount of protein was further analyzed by sucrose gradient sedimentation, capture enzyme-linked immunosorbent assay (ELISA), and electron microscopy. Further details are provided as Supplementary Data (Supplementary Data are available online at
Structural characterization of fusion proteins
Sucrose gradient sedimentation analysis
Five micrograms of fusion proteins were layered onto a linear gradient of 5% to 50% sucrose, and centrifuged at 36,000 rpm, 3 h, 4°C, without brake, using a SW41 rotor (Beckman). Fractions (0.7 mL each) were collected and pellets were suspended in 500 μL of phosphate-buffered saline (PBS). HPV 16 L1 VLPs [150S (30)], L1( [10S (58)] capsomers and catalase (11S) were used as controls. Fractions were analyzed by WB using an anti-HPV 16 L1 monoclonal antibody.
Transmission electron microscopy
Samples (3 μL with 0.03 μg/μL of each chimeric protein) were loaded onto carbon-coated grids, then negatively stained with 2% uranyl acetate, and analyzed with a transmission electron microscope CM200 FEG (FEI) operating at 200 kV.
ELISA with HPV 16 L1-specific antibodies
Purified proteins (0.5 μg/well diluted in 50 μL PBS) were directly coated on 96-well plate (Nunc) and incubated overnight at 4°C. After washing four times with 200 μL per well of PBS-0.05% Tween 20; 100 μL per well of blocking buffer (5% milk in PBS-0.05% Tween 20) were applied and incubated for 1 h at 37°C. Then, plates were loaded with 50 μL/well from different HPV 16 L1-specific monoclonal antibodies (1:1,000 in blocking buffer): H16.V5, H16.U4, MD2H11, and CamVir-1; the first two against conformational L1 epitopes, and the last two against linear L1 epitopes. After 1 h at 37°C, plates were washed four times, and 50 μL per well of HRP-conjugated goat anti-mouse IgG antibody (Dianova) at 1:1,000 dilution was added to each well. After another hour at 37°C, the plate was washed again, and 50 μL per well of ABTS (2,2′-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium; Sigma) substrate were added.
The absorption was measured in an ELISA reader at 405 nm. Data presented are means of duplicates with standard error of the mean (SEM).
Upscaling of production and purification of L1Δ+E2H/NC chimeric capsomers
To obtain larger quantities of the L1Δ+E2H/NC protein for in vivo analysis, JM109-DE3 bacteria were transformed with the plasmid carrying L1Δ+E2H/NC and cultured in LB medium supplemented with 100 μg/mL ampicillin at 30°C, 200 rpm. When the culture reached an optical density of 0.4 at 600 nm, IPTG at 0.2 mM final concentration was added; and after 2 h of incubation, bacteria were harvested and suspended in buffer L with 2 mM of DTT, 50 μg/mL lysozyme, and complete protease inhibitor cocktail; and lysed by sonication.
Purification was performed using a Glutathione Sepharose™ 4B system (17-0756-01; GE Healthcare), as detailed in Supplementary Data, using 80 U/mL of thrombin protease to cleave L1Δ+E2H/NC from GST. Lipopolysaccharides (LPS) were removed by treatment with 1% Triton X-114 (Sigma) (43). Protein purity and concentration were determined by comparing to a bovine serum albumin (BSA) standard on a Coomassie-stained SDS-PAGE. Proteins were sterilized by λ radiation (9,000 rads) using a 137Cs source.
Construction of pCDNA4/TO-E2HPV 16 vector
The full-length HPV 16 E2 gene was cloned into pCDNA4/TO vector under the control of the cytomegalovirus promoter, using BamHI and EcoRI restriction sites. The E2 gene was obtained from plasmid pGex-4T2 containing L1Δ+E2full through PCR. The resulting vector pCDNA4/TO-E2HPV 16 was linearized with BcgI restriction enzyme and used to transfect the TC-1 cell line.
Cell lines
RMA-S (28), a transporter associated with antigen processing (TAP)-deficient mutant derived from RMA cell line (H2-Db), and K562 (ATCC®CCL-243™), a highly sensitive target for natural killer cells, were cultured in RPMI medium supplemented with 10% inactivated FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin. TC-1 cell line, derived from C57BL/6 primary lung epithelial cells and transformed by HPV 16 E6/E7 and c-Ha-ras (27), was cultured in DMEM with 7% FBS, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 400 μg/mL of geneticin. TC-1/E2 cells expressing HPV 16 E2 protein were produced by stable transfection of TC-1 cells using pCDNA4/TO-E2HPV16 vector and selected with 200 μg/mL of zeocin; TC-1/TO− cells were transfected with pCDNA4/TO. All cell cultures were grown at 37°C, in a humidified atmosphere of 5% CO2.
Mice
Ten to 12-week-old C57BL/6 female mice were kept at the animal facilities from the Biomedical Research Institute (UNAM-México) under specific pathogen-free conditions, in compliance with institutionally approved protocols and according to Official Mexican Norm NOM 062-ZOO-1999.
Immunization
Mice were immunized intraperitoneally with 5 μg of fusion protein L1Δ+E2H/NC diluted in 50 μL of PBS and emulsified with 50 μL of Freund's adjuvant; 10 days later they receive a second equal dose of antigen and after 15 days, the third. L1Δ+E2H/NC or PBS, as control, was mixed with same volume of complete Freund's adjuvant (Sigma) for the first immunization, and with incomplete Freund's adjuvant (Sigma) for the latter two. Another control group was immunized with 5 μg/mL of HPV16 L1 protein (as commercial vaccine Gardasil®) with the same schedule.
In vitro restimulation of splenocytes and cytotoxicity assays
Mice were immunized with L1Δ+E2H/NC protein, HPV16 L1, or PBS; their spleens were removed and pooled to prepare a single cell suspension. Erythrocytes were lysed and splenocytes were cultured in enriched IMDM medium supplemented with 30 ng/mL of IL-2 (R & D systems) and 1 μg/mL of L1Δ+E2H/NC protein, 1 μg/mL of HPV16 L1 (Gardasil) or without antigen, respectively.
51Cr release cytotoxicity assay. On day 10, cells were harvested and cocultured during 2 h at 37°C, 5% CO2 with K562 cells; then, titrated effector cells were mixed with 5,000 target cells per well, and incubated during 4 h at 37°C. TC-1/E2 and TC-1/TO-, or RMA-S cells loaded with different E2 peptides, were pulsed with Na2 51CrO4, and used as target cells. Released radioactivity was measured on a γ-counter (Packard). The percentage of specific lysis was calculated as follows: (experimental release–spontaneous release)/(maximum release–spontaneous release) × 100; and presented as the means from triplicate wells with SEM. See Supplementary Data for details.
Cytotoxicity assay using cytometry with carboxyfluorescein succinimidil ester (CFSE) and 7-amino-actinomicin (7AAD). Restimulated cells were harvested on day 8; CD8+ cells were enriched (EasySep Mouse CD8+ T Cell Isolation Kit, 19853; StemCell) from splenocytes, titrated (2 and 1 × 105) and challenged against 5 × 104 TC-1/E2 or TC-1/TO− target cells labeled with CFSE, in a total volume of 200 μL in round-bottom 5-mL tubes. After incubating 4 h at 37°C, cells were harvested, washed, and stained with 7AAD (Via-Probe Cell Viability Solution, 555816; BD Bioscience). Finally, cells were counted by flow cytometry (FACSCalibur; BD Bioscience), and data were analyzed using Summit V4.3 software.
Tumor inhibition assay
C57BL/6 mice were injected subcutaneously in the dorsum with 60,000 TC-1/E2 cells. Once tumors reached a volume of 20–30 mm3, the group was divided in two, and the immunization schedule started as indicated earlier. A group of mice was immunized with L1Δ+E2H/NC protein, and the other received PBS with adjuvant. Tumor size was measured with an electronic caliper during the indicated days from first immunization. Tumor volume was calculated as V = (a × b × c)/2, where (a) is the length, (b), width, and (c) height of the tumor (47). Data presented are the means of the specified number of mice with SEM. Differences between the groups were evaluated by two-tailed Fisher's exact test using GraphPad Prism 6 software. All p-values below 0.05 were considered statistically significant.
Epitope prediction
Three algorithms were used to predict putative epitopes from HPV 16 E2H/NC (aa 206–307), which could bind to H2-Db molecule: BIMAS (36), SYFPEITHI (38) and RANKPEP (39). Custom purchased peptides were synthetized by solid phase peptide synthesis (Invitrogen) and purified by high performance liquid chromatography (HPLC; Invitrogen).
RMA-S binding assay
RMA-S cells (2 × 105) were incubated overnight at 37°C with titrated concentrations (0.8 to 50 μM) of peptides in a 96-well U-bottom plate. Then, 100 μL of anti H-2Db hybridoma supernatant 28-8-6S (ATCC HB-51™) were added, and incubated 30 min on ice; after washing, plate was incubated for 30 min on ice with fluorescein isothiocyanate-conjugated (FITC) goat anti-mouse antibody. Stained cells were analyzed by flow cytometry and mean fluorescence intensities (MFI) were recorded to calculate the fluorescence index (FI) as follows: FI = (MFI with peptide/MFI without peptide).
Results
HPV 16 L1Δ+E2 chimeric proteins form heterogeneous particles
As indicated in Figure 1, we designed and produced in E. coli six chimeric proteins with a modified version of L1 from HPV 16, fused to the full-length E2 gene (L1Δ+E2full) or to a fragment containing the N1-terminal subdomain (L1Δ+E2N1), the N2-terminal subdomain together with a short hinge region (L1Δ+E2N2), the N2 terminal subdomain linked to a more extended hinge region spanning to aminoacid 260 (L1Δ+E2N2/H), the complete C-terminal domain, with a short hinge region (L1Δ+E2C), and a partial C-terminal domain with the hinge region (L1Δ+E2H/NC).
First, we wanted to assess whether the three-dimensional structure acquired by the chimeric proteins allows the formation of capsomers. To this end, we performed size exclusion chromatography. The chromatogram showed the peak of total protein at fraction #4 (not shown), which also contained the highest amount of HPV 16 L1, as detected by WB analysis using an anti-L1 monoclonal antibody (Fig. 2a). This result indicated that chimeric proteins adopt an arrangement that corresponds to an MM (molecular mass) of 2,000 kDa and higher. Since this is more than the molecular mass of capsomers (260 kDa), we speculate that we have heterogeneous polymers formed by ≥30 monomers.

Structural characterization of chimeric proteins. After production in Escherichia coli and purification by GST-affinity chromatography, proteins were analyzed by size exclusion chromatography,
Additionally, to extend the structural analysis of the chimeric proteins, we used the fraction #4 of the size exclusion chromatography from the different proteins to analyze their sedimentation behavior through sucrose gradients (5–50%). The fractions obtained after centrifugation were analyzed by WB (Fig. 2b). Except for the L1Δ+E2 full protein, we found that a large amount of chimeric proteins sediment in fractions 10–15. However, we also see them in fractions 5–7, which correspond to VLPs, and in fractions 14–17, which correspond to individual capsomers.
These results agree with those obtained by size-exclusion chromatography: the chimeric proteins form a heterogeneous mix of different-sized particles containing between 1 capsomer and 72 capsomers (VLPs). To further assess the conformation of our proteins we used electron microscopy. Fractions containing the different chimeric proteins were stained with 2% uranyl acetate and analyzed. As shown in Figure 2c we can indeed observe different sizes of particles.
The chimeric protein L1Δ+E2H/NC retains the HPV 16 L1 neutralizing epitope
Next, we wanted to know whether these heterogeneous particles still maintained the L1-specific neutralizing epitopes. We performed an ELISA with different antibodies against L1 from HPV 16 identifying either linear (Camvir-1, MD2H11) or conformational epitopes (H16.V5, H16.U4). H16.V5 recognizes a neutralizing epitope against L1 (53).
As can be seen in Figure 3, our six fusion proteins exhibit a positive reaction when evaluated with the antibody Camvir-1 and to a lesser extent exhibit a positive response to the MD2H11 antibody. However, concerning the neutralizing epitope recognized by H16.V5 antibody, only L1Δ+E2H/NC and L1Δ+E2C proteins showed a positive result, with a much higher reactivity by the former. The reactivity of L1Δ+E2H/NC protein to the H16.V5 antibody strongly suggests that the protein could be capable of eliciting the production of neutralizing antibodies, and hence of inducing a prophylactic response in immunized recipients. Based on this result, we decided to analyze cellular immunogenicity only with L1Δ+E2H/NC.
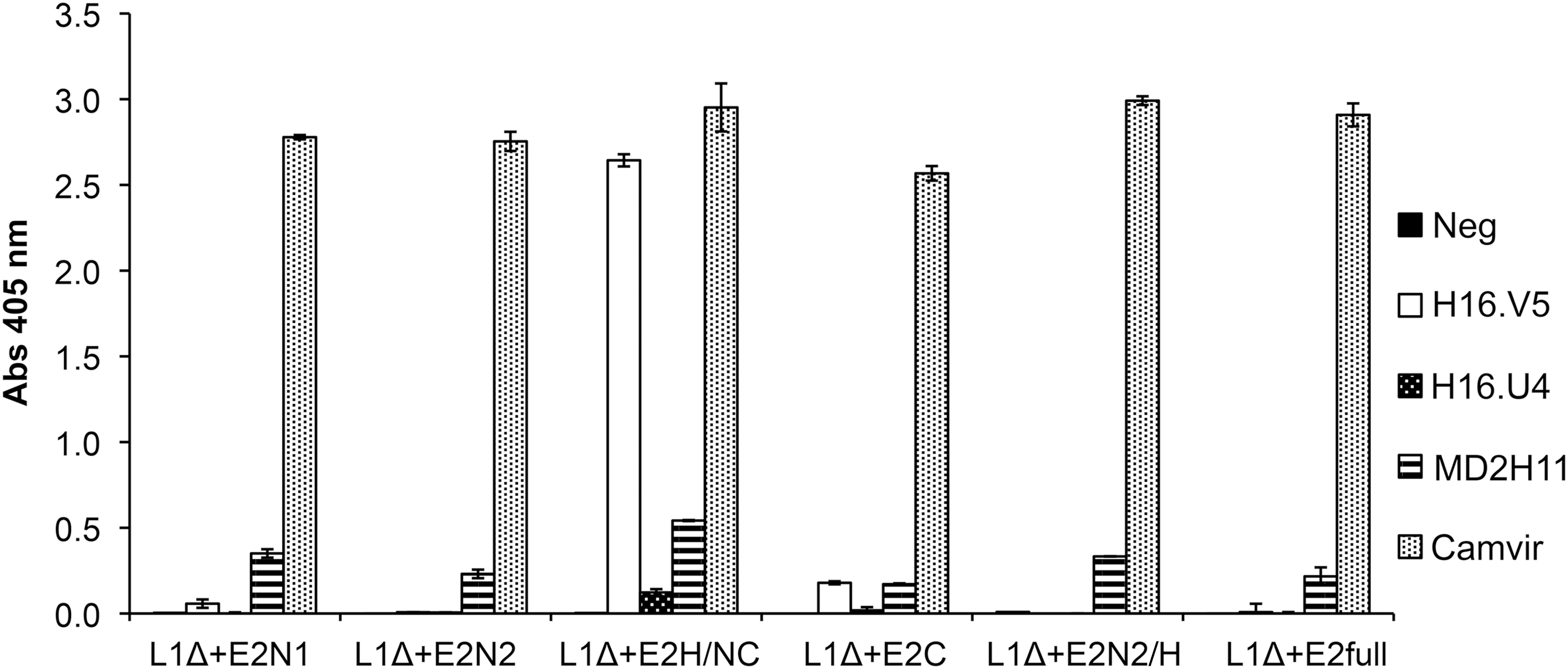
Presence of L1 epitopes within chimeric proteins was determined by enzyme-linked immunosorbent assay using different anti-L1 monoclonal antibodies. Proteins obtained from size exclusion chromatography were analyzed using antibodies that recognize linear (MD2H11, Camvir) and conformational (H16.V5, H16.U4) epitopes on L1.
To obtain enough protein for immunization, this protein was produced at a larger scale in E. coli JM109-DE3 (as detailed in the Materials and Methods section). After harvesting and lysing of the bacteria, the clear supernatant was incubated with Sepharose 4B beads to allow binding of L1Δ+E2H/NC protein, and the flow-through was collected (FT, Fig. 4a). Beads were later washed four times (W1, first wash; W4, fourth wash, Fig. 4a) with buffer L, before incubation with thrombin. We eluted a 63 kDa protein corresponding to L1Δ+E2H/NC (ET, Fig. 4a) now separated from the GST tag. After one more wash step (WT, Fig. 4a), beads were incubated with reduced glutathione to detach the GST tag and check the efficiency of thrombin digestion. We eluted a clean band of 26 kDa corresponding to GST (EG, Fig. 4a), instead of the 90 kDa band of complete GST+ L1Δ+E2H/NC protein. The last wash was performed to remove all proteins (WG, Fig. 4a).
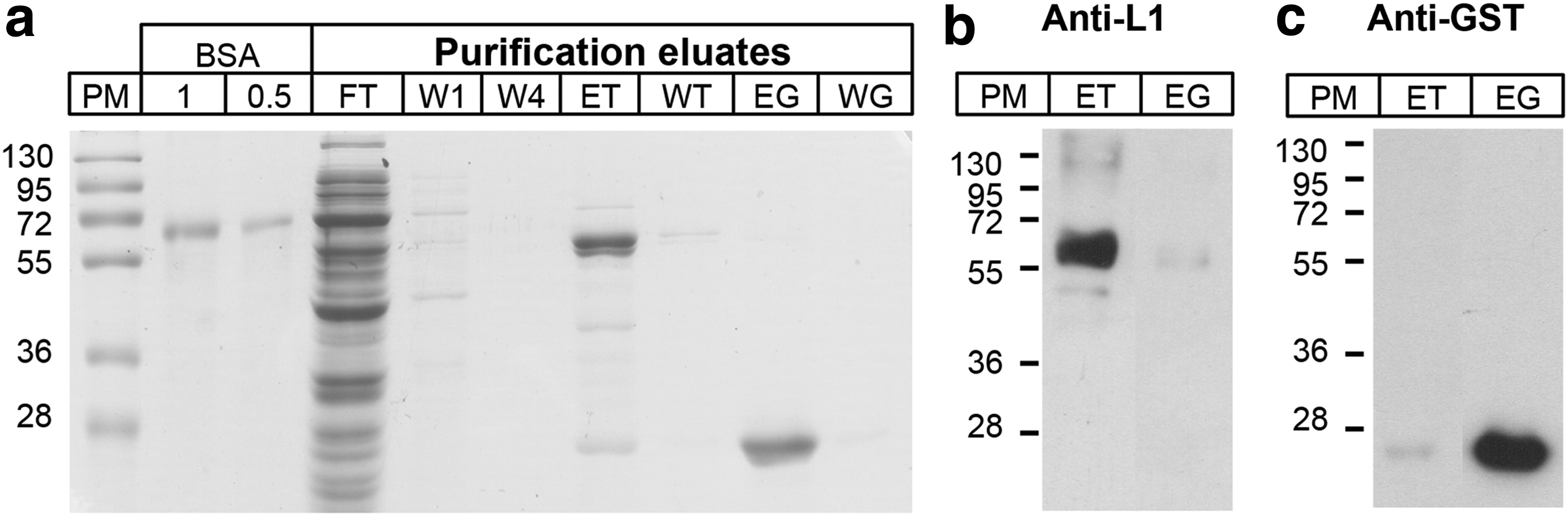
Purification of L1Δ+E2H/NC chimeric proteins from E. coli JM109-DE3 cultures by GST-glutathione affinity chromatography.
The L1Δ+E2H/NC protein (ET eluate) was treated with phenylmethylsulfonyl fluoride (PMSF) and Triton X-114 to remove LPS. It was then quantified by comparing with known amounts of BSA (67 kDa) in a 12% polyacrylamide gel stained with Coomassie blue, and using Image J software. To corroborate that the bands observed in PAGE corresponded to L1Δ+E2H/NC (Fig. 4b) and GST (Fig. 4c) we performed WBs with the respective antibodies (anti-L1 HPV 16, and anti-GST-HRP). We obtained L1Δ+E2H/NC protein at the concentration of 0.36 μg/μL, yield of 270 μg/L, and purity of 97%. Protein was sterilized by radiation as described in the Materials and Methods section, to be used as an immunogen.
Immunization with L1Δ+E2H/NC chimeric protein elicits cytotoxic cellular response against HPV 16 E2
We next evaluated the capacity of L1Δ+E2H/NC to induce a cellular immune response by two different cytotoxicity assays: 51Cr release assay and flow cytometry using CFSE/7AAD. We adapted a murine model using C57BL/6 mice and the syngeneic TC-1 cell line. TC-1/E2 cells were established transfecting TC-1 cells with pCDNA4/TO-E2HPV16 vector (as described in Materials and Methods). Presence of the E2 gene was corroborated by PCR (not shown). WB analysis with an E2-specific monoclonal antibody confirmed the expression of a 43 kDa band in cells transfected with the pCDNA4/TO-E2HPV16 vector, whereas it was not seen in those cells transfected with the empty vector (not shown). These E2-expressing TC-1 cells were used as target in the cytotoxicity assays.
Three female C57BL/6 mice were immunized intraperitoneally three times with 5 μg of L1Δ+E2H/NC protein (as detailed in Materials and Methods) emulsified in Freund's adjuvant. After three weeks, mice were sacrificed; their splenocytes were removed, pooled, and restimulated ex-vivo. After 10 days, effector cells were challenged against target TC-1/E2 and TC-1/TO cells previously labeled with 51Cr. After 4 h of incubation, liberation of chromium into the supernatants was measured. We observed that the splenocytes had a cytotoxicity of about 50% against the E2-expressing cells and almost none against the cells which do not (Fig. 5a).
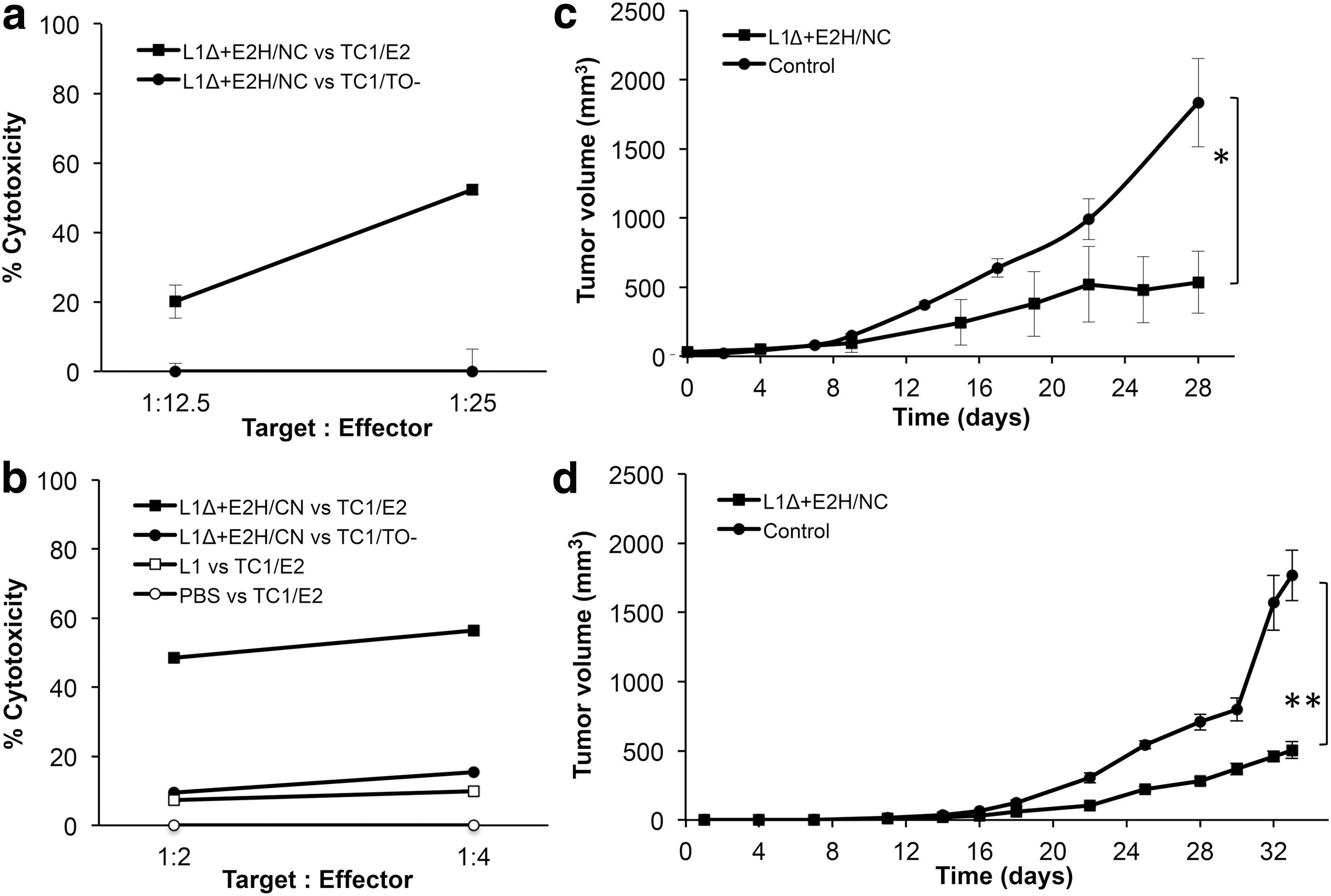
Specific cytotoxic activity and antitumoral effect of CTLs induced by immunization with L1Δ+E2H/NC protein on C57BL/6 mice.
The cytotoxicity of lymphocytes was also assessed by flow cytometry using CFSE/7AAD. Three groups of mice were immunized: one group received 5 μg of L1Δ+E2H/NC protein in Freund's adjuvant (n = 4), another group 5 μg of HPV16 L1 protein (Gardasil®; n = 3), and the third received PBS in adjuvant (n = 3). Three weeks after last immunization, mice were sacrificed; their spleens were removed and splenocytes were restimulated ex-vivo during 7 days; then CD8+ lymphocytes were enriched and challenged against target cells in different proportions. Target cells were previously stained with CFSE; then cocultured with CD8+ T cells; and after 4 h, cells were harvested and stained with 7AAD.
As shown in Figure 5b, the cytotoxic CD8+ lymphocytes obtained from mice immunized with L1Δ+E2H/NC protein, lysed 56% of E2 protein expressing target cells (TC1/E2) while they lysed TC1/TO− cells that do not express the E2 protein far less. This cytotoxicity was not observed from CD8+ lymphocytes obtained from mice immunized with L1 alone (as Gardasil®) or with PBS in adjuvant, when they were challenged against the same TC-1 cells expressing E2 (TC-1/E2).
These results show that immunization with L1Δ+E2H/NC chimeric protein induces cytotoxic cellular responses capable of recognizing and lysing specifically E2 expressing cells.
Immunization with L1Δ+E2H/NC chimeric protein inhibits tumor growth in mice
We wanted to test whether the E2-specific cellular response induced by immunization with L1Δ+E2H/NC protein affects the growth of tumors in vivo. To this end, we performed tumor rejection experiments using TC-1/E2 cells.
Thirteen C57BL/6 mice received 60,000 TC-1/E2 cells s.c. on their dorsum. Once the tumors reached a volume of 20–30 mm3, immunization was started (as detailed in Materials and Methods). A group of seven mice were immunized with L1Δ+E2H/NC protein emulsified in Freund's adjuvant while another group of six mice received PBS in adjuvant. The tumor volume was measured over a 33-day period. At this point, mice immunized with L1Δ+E2H/NC had an average tumor volume of 505 mm3, whereas control group had 1,776 mm3 (Fig. 5d); although tumor growth was not completely ablated, the immunization with L1Δ+E2H/NC inhibited tumor growth by 70%, and this difference was statistically significant (p = 0.0210).
Another independent experiment was done, this time four mice were immunized with L1Δ+E2H/NC and another four, with PBS in adjuvant. At day 28, the former group had an average tumor volume of 535 mm3, whereas control group had 1,835 mm3 (Fig. 5c). It was also observed that ∼70% of tumor growth was inhibited (p = 0.0286). In addition, another group of three animals was immunized with L1 from HPV16 (Gardasil®) and at day 31 found to have had an average tumor volume of 1,559 mm3, similar to that of animals immunized with PBS (not shown).
From all these data, we conclude that immunization with L1Δ+E2H/NC chimeric protein induces a cytotoxic T-cell response against HPV 16 E2 and inhibits tumor growth in mice.
Identification of an epitope of HPV 16 E2 involved in the induction of cytotoxic response
Finally, we wanted to define the epitopes of E2 within the L1Δ+E2H/NC chimeric protein (E2 aa 206-aa 307). We screened for peptides that fit the MHC I-H2-Db molecule on the primary structure of E2 protein. Using epitope prediction softwares, we found five peptides with probable affinity to H2-Db molecules, that is, (i) peptide E2-206 (including aminoacids 206 to 215 of HPV 16 E2 protein, with the sequence SSPEIIRQHL), (ii) peptide E2-213 (aa 213–221, QHLANHPAA), (iii) peptide E2-243 (aa 243–252, SEPDTGNPCH), (iv) peptide E2-280 (aa 280–288, NCNSNTTPI), and (v) peptide E2-292 (aa 292–301, KGDANTLKCL). The in vitro affinity of these peptides was then evaluated using a RMA-S binding assay. Peptide E2-292 showed the high affinity to murine MHC-I molecules H2-Db, as detected by fluorescence-activated flow cytometry, while the rest do so only when present at high concentrations (50 μM; Fig. 6a). This indicates that peptide E2-292 constitutes a possible epitope for the induction of the E2-specific cellular response seen in immunized mice. To address this, we tested whether the splenocytes obtained from mice immunized with L1Δ+E2H/NC protein would be capable of recognizing the E2 peptides on the surface of target cells.
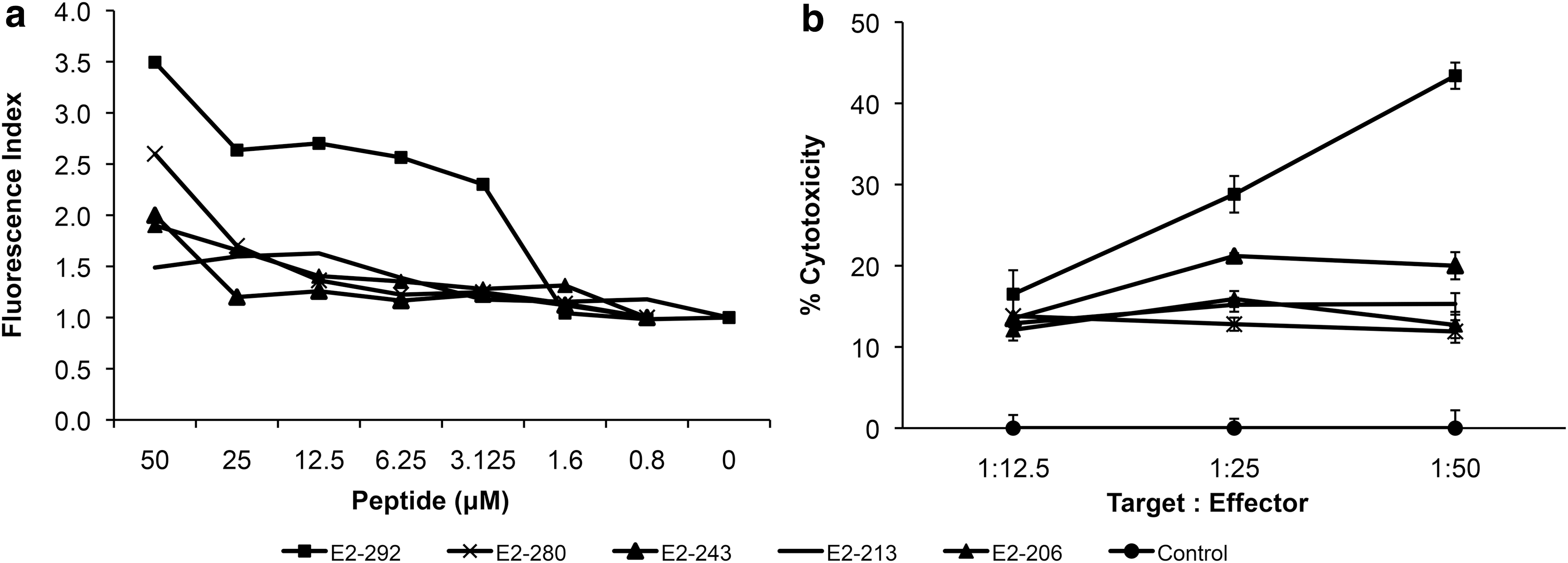
Binding of HPV 16 E2 synthetic peptides to H2-Db molecule on RMA-S cells and their recognition by splenocytes derived from L1Δ+E2H/NC protein-immunized mice.
C57BL/6 mice were immunized three times with 5 μg of L1Δ+E2H/NC protein (same protocol used above), their splenocytes were restimulated and then loaded on a 96-well microplate, together with target cells in 51Cr release assays. RMA-S cells loaded with each peptide at a concentration of 50 μM and labeled with 51Cr were used as target cells. Liberation of chromium into the supernatants was measured. While splenocytes challenged against RMA-S cells loaded with E2-206, -213, -243, or -280 peptides had poor cytotoxic activity (in line with their weak affinity for H2-Db molecule), splenocytes challenged against RMA-S cells loaded with E2-292 peptide, showed high cytotoxicity (Fig. 6b), similar to that obtained against target cells expressing the whole E2 protein.
These results show that the E2-292 peptide is an important H2-Db-restricted epitope involved in the cytotoxic cellular response in mice against cells and tumors positive for HPV 16 E2.
Discussion
The burden of cervical cancer is largely associated with poor access to healthcare systems, and although current HPV L1 VLP-based prophylactic vaccines prevent infection by major HR-HPV types, they do not help women with already established infections and without detectable lesions that, together with poor coverage of early cancer detection and/or HPV screening programs in developing countries, renders them at a high risk to develop cervical lesions and cancer. Therefore, there is a requirement for a therapeutic vaccine that is preferably also endowed with prophylactic properties to prevent reinfection by the same HPV type. Chimeric capsomers have been proposed to fulfill these properties.
Previously, it has been shown that capsomers of L1 protein induce neutralizing antibodies (16,41,42,46) that can prevent papillomavirus infection (56,58) and can also be processed through MHCI pathway to induce (against L1 as proof of principle) cellular responses and inhibit tumor progression in mice (35).
In addition, capsomers of L1 can be produced in bacteria, which offers a less costly process and yield thermostable proteins, while conserving their structure (6,7) and their ability to generate neutralizing antibodies (56) similar to those produced in insect cells (43). The risk incurred through use of LPS can be reduced by efficiently removing them with Triton X-114 (43). Indeed, recently, a bivalent HPV 16/18 prophylactic vaccine produced in E. coli. has proved to be safe and immunogenic in a phase 2 clinical trial (55). In addition, chimeric capsomers also have been shown to conserve their immunogenicity when fused to another protein (E7 protein from HPV 16) (1).
Cellular immune response generated against E2 has shown potential therapeutic effects in animal models (22,44,45), and it also appears to be involved in human cellular responses against E2 (2,10 –12,21,25,48,49,54).
E2 protein from HPV 16 has been used as protein antigen in mice, showing the induction of E2-specific lymphoproliferation and cytotoxicity in vitro (20), although the effect on tumors was not assessed. Other groups have used vaccinia virus Ankara (17) and adenovirus vectors (50), despite the concomitant risk of using virus. A chimeric construct including E2 has also been generated, however, it was produced in insect cells, and its in vivo effect was not tested (37).
The cellular response against E2 would be beneficial to women with low-grade cervical lesions, since it is expressed in intermediate cell layers of the stratified cervical epithelium (57), and, together with the E1 protein is necessary to establish an infection and maintain viral episomes in basal layer cells (14); in addition, it would help eliminate incipient and latent infections along with low-grade cervical lesions that persist or recur after treatment.
Here, we aimed to generate chimeric capsomers of L1 proteins carrying different parts of E2. We used a modified version of L1Δ (L1ΔN10ΔC29), which has been shown to be more soluble in bacteria due to the lack of 10 residues from the amino-terminal domain (6), maintaining the structure and immunogenicity of the full-length L1 from HPV 16 (42). Full-length L1 protein is a “jelly-roll” β-sandwich with a core of 10 β-strands and five loops in the apical region, a bottom region with five α-helices laterally projected that contacts an L1 protein in the neighboring capsomer, and a tail sequence that is disordered and flexible; deletion of 29 aa of this carboxy-terminus neither affects the stability of the protein nor the formation of L1 capsomers (7,34), and can be substituted by another protein as carried antigen, as in this case was substituted by E2 fragments. This construction (L1ΔN10ΔC29) was characterized by Schädlich, et al. (42).
We produced chimeric proteins fusing L1ΔN10ΔC29 with E2 full-length protein and with segments of it. The construct L1ΔN10ΔC29+E2H/NC conserved the H16.V5 epitope, which is necessary and sufficient for induction of neutralizing antibodies (53), and the most abundant epitope for neutralizing antibodies seen in HPV-vaccinated subjects (51). The fact that this construct exhibits the H16.V5 epitope reinforces the idea that this chimeric protein is assembled in capsomers, since H16.V5 antibody binds to an epitope covering an arrangement of 17 aa across five intertwined loops in the apical immunogenic surface of two neighboring L1 proteins within a capsomer (26).
Although we suggest the fusion protein L1ΔN10ΔC29+E2H/NC as potential prophylactic vaccine, it would be mandatory to assess its capacity to indeed induce neutralizing antibodies in vivo. We speculate that this construct is the only one bound by the H16.V5 antibody due to the structure of the E2H/NC segment, where 28 aa from the carboxy-terminus are preceded by the hinge region, a known flexible aminoacid sequence (29); while the other fusion proteins carry sequences from the amino or carboxi domains of E2, whose native structures are well structured. The amino-terminus is an activation domain consisting of two subdomains, an anti-parallel β-sheet and an helical subdomain; while the carboxy-terminus has a β-sheet half-barrel structure (19); this could destabilize the structure of L1 protein, rendering it only available to antibodies recognizing linear epitopes.
The very low reactivity of the H16.U4 antibody to the fusion protein L1Δ+E2H/NC observed in our assay, could be due to the location of the epitope (aa 427–445), in the carboxy-terminal protruding arm of L1, which forms the intercapsomeric cleft in VLPs; this conformational epitope is located between the helix 4, which contacts the neighboring L1 protein, and the short βJ strand; then, this is followed by the helix 5 (4,33) and, instead of the last disordered 31 aa of L1, the E2H/NC segment protein. We speculate that this protein is disturbing or obstructing the H16.U4 epitope, which together with the low affinity constant of the antibody, which is 10 times lower than that of H16.V5 (9,52), results in fewer antibody binding.
It appears that E2H/NC is disturbing the protruding arm of L1, but not its more tightly structured core, in which on top is located the H16.V5 epitope (5,8). The neutralizing capacity of H16.U4 antibody is weaker than that of H16.V5 (9,40); it has been shown that the lack of the epitope does not avoid the capacity of the particle to induce neutralizing humoral response, as it does in the case of H16.V5 (53).
On the other hand, when we tested the effect on cellular immune response of immunization with L1ΔN10ΔC29+E2H/NC in mice, we observed that this chimeric protein was capable of activating CD8+ cytotoxic lymphocytes against tumor cells expressing E2 in a 51Cr release cytotoxic assay and in a CFSE/7AAD flow cytometry assay. The cytotoxicity detected by using the former assay was 52%, and by using CFSE/7AAD was 56%, even though distinct proportions of target:efector cells were used (1:25 and 1:4, respectively). This could be explained by the use of enriched/purified CD8+ T lymphocytes in CFSE/7AAD experiments, whereas in the 51Cr release cytotoxic assay the bulk of splenocytes was used.
Most importantly, this specific cytotoxic activity of CTLs induced by immunization with L1Δ+E2H/NC protein can counteract by 70% the growth of E2-expressing tumors in C57BL/6 mice with already developed tumors; this context resembles that found in women with existing lesions, although this animal model has limitations, since the tumor was subcutaneously injected in the dorsum of mice, and not generated in the cervix.
Then, we propose that the activated T-cell population induced by immunization with L1Δ+E2H/NC consists, in a high percentage, of T-cell clones specific for the peptide E2 292–301, since the cytotoxicity assay showed higher level of target cell lyses when cells were loaded with this peptide. In addition, this peptide is contained in the carboxy-terminal region of E2, which has been shown to be the most immunogenic among the entire protein (2,20).
Thus, we conclude that immunization with chimeric L1Δ+E2H/NC protein obtained in bacteria induces a specific cytotoxic response against the E2 protein from HPV 16, which is capable of inhibiting tumor growth in mice, exhibiting therapeutic activity. In addition, this chimeric protein could potentially have prophylactic activity, since a major neutralizing epitope of L1 from HPV 16 is maintained. Finally, we found that this epitope from E2 (aa 292–301) induces strong cytotoxic activity and propose that it should be tested whether it can also induce strong cytotoxic activity in humans and so could be included in the generation of new therapeutic vaccines against HPV.
Compliance with Ethical Standards
Author's contribution: L.G. and A.G.C. conceived and designed the study, G.L.T. performed the experiments, generated, analyzed, and interpreted data; L.S. participated in the construction, expression and purification of recombinant clones, and ELISA experiments; A.J.A.-C. and M.C.G. participated in the acquisition of data from experiments with mice; M.C.G. participated in the construction and expression of E2 for animal experiments; R.G.R. and A.M.-G. helped perform experiments with 51Cr and data acquisition; G.L.T. and A.G.C. drafted the article and corrected revised versions. All authors have approved the final article.
Footnotes
Acknowledgments
We acknowledge EM de Villiers and H zur Hausen for their kind gift of complete HPV 16 genome cloned in pBR322; V. Ortiz Navarrete for kindly providing anti H-2Db hybridoma supernatant 28-8-6S (ATCC HB-51); and for kindly donating the RMA-S cell line, and N. D. Christensen for kindly providing primary antibodies H16.V5, H16.U4, and MD2H11; and E. Langley McCarron, for her profuse help in English language editing We acknowledge generous support from National Council of Research and Science (Consejo Nacional de Ciencia y Tecnología [CONACYT], México, grant number 127722, to A.G.C.), and Agreement México-Germany (J110.530/260, Instituto Nacional de Cancerología-DLR, 2007–2008). Funding sources had no involvement in any aspect of this research. Gabriela López Toledo, a PhD student in the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México, received a scholarship (#199976) from CONACYT, México.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.