
Abstract
Human respiratory syncytial virus (HRSV) infections have worldwide records. The virus is responsible for bronchiolitis, pneumonia, and asthma in humans of different age groups. Premature infants, young children, and immunocompromised individuals are prone to severe HRSV infection that may lead to death. Based on worldwide estimations, millions of cases were reported in both developed and developing countries. In fact, HRSV symptoms develop mainly as a result of host immune response. Due to inability to establish long lasting adaptive immunity, HRSV infection is recurrent and hence impairs vaccine development. Once HRSV attached to the airway epithelia, interaction with the host innate immune components starts. HRSV interaction with pulmonary innate defenses is crucial in determining the disease outcome. Infection of alveolar epithelial cells triggers a cascade of events that lead to recruitment and activation of leukocyte populations. HRSV clearance is mediated by a number of innate leukocytes, including macrophages, natural killer cells, eosinophils, dendritic cells, and neutrophils. Regulation of these cells is mediated by cytokines, chemokines, and other immune mediators. Although the innate immune system helps to clear HRSV infection, it participates in disease progression such as bronchiolitis and asthma. Resolving the mechanisms by which HRSV induces pathogenesis, different possible interactions between the virus and immune components, and immune cells interplay are essential for developing new effective vaccines. Therefore, the current review focuses on how the pulmonary innate defenses mediate HRSV clearance and to what extent they participate in disease progression. In addition, immune responses associated with HRSV vaccines will be discussed.
Introduction
H
In infants, where the adaptive immune system is premature, the innate immune system plays a central role in the defense against HRSV. Immune response to HRSV infection begins in the upper respiratory tract where it interacts with pulmonary innate immunity such as epithelial cells, mucus, neutrophils, natural killer (NK) cells, macrophages, and dendritic cells (DCs) and their immune mediators (Fig. 1) (49,273). These immune mediators drive a massive leukocyte infiltration into the lungs, which deposits on the epithelial cells, resulting in their death (4,65,77,174,130). Dead epithelial cells along with mucus form thick plugs that obstruct airways leading to bronchiolitis (188,244). Established bronchiolitis symptoms involve wheezing, dyspnea, tachypnea, and hypoxia (212).
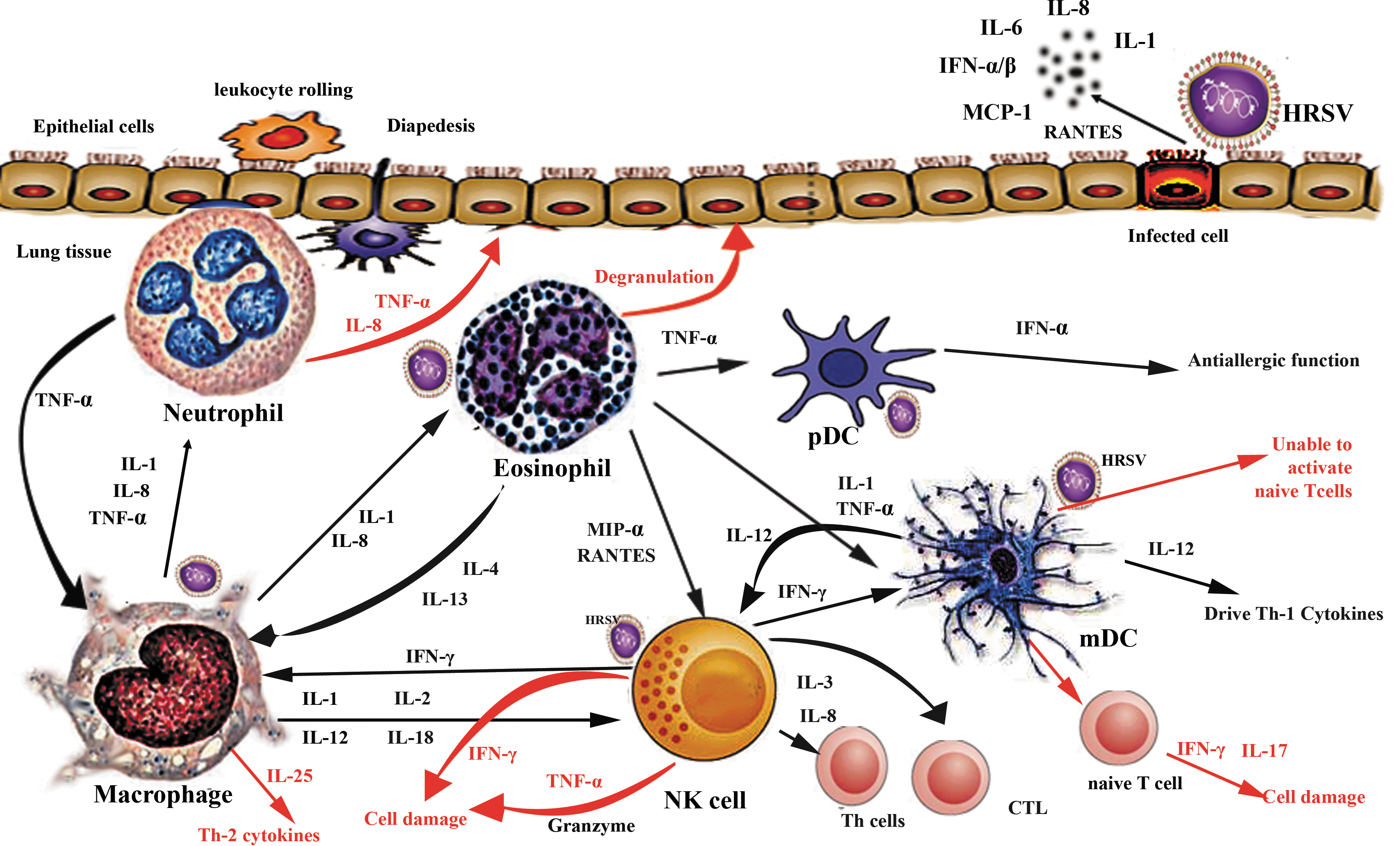
A schematic representation showing how innate immune cells participate in virus clearance and disease progression during HRSV infection. The figure describes virus–cell, cell–cell interaction, and subsequent release of immune mediators. Innate immune response is initiated once the virus infects the airway epithelia (the primary target), which respond to viral infection by secreting numerous immune mediators. These mediators act to recruit other leukocytes to the site of infection, which in turn release their own cytokines that help to amplify the immune response. Black arrows refer to possible interactions between innate immune cells during HRSV infection. Red arrows refer to the contribution of innate immune cells/mediators in disease enhancement. HRSV, human respiratory syncytial virus. Color images available online at
The subsequent sections will discuss the following points: (i) the role of pulmonary innate leukocytes in terms of their recruitment and activation, (ii) cytokines involved in initiation of the innate immune response as well as their role in disease augmentation, (iii) different HRSV vaccines and their protective efficiency.
The Role of Innate Immune Components in HRSV Clearance and Disease Progression
Epithelial cells
Epithelial cells represent the primary target for HRSV infection and replication. The virus structural proteins, G and F, interact with different epithelial cell receptors such as glycosaminoglycan and C-type lectins. In addition, they also interact with the immune receptors CX3CR1 and Toll-like receptor (TLR)-4, respectively, and directly affect the epithelial cell response in terms of secretion of inflammatory cytokines and release of oxygen radicals (200,253). Epithelial cells of the airway orchestrate the innate immune response through a group of cytokines and growth factors that regulate recruitment, activation, and differentiation of several leukocytes.
These mediators involve IL-1, IL-6, IL-11, CXCL10, IL-8, CCL-5, MCP-1, MIP-1α, TNF-α, IFN-α/β, and GM-CSF and their expression is upregulated during HRSV infection (Fig. 1) (66,80,180,201,214,218,257,270). Upregulating the expression of such mediators is initiated by activation of NF-κB signaling cascade (8,18,106,113). In addition, epithelial cells facilitate phagocytosis by secretion of opsonizing molecules such as complement and surfactant proteins (collectin, SP-A) (127,271). These proteins were shown to facilitate HRSV clearance both in vitro and in mice models (83,103,139).
Mucus, another important innate defense factor, is secreted by specialized epithelial goblet cells. It forms a thick gelatinous layer that covers the airway epithelia. It protects respiratory airways from injury due to its viscosity, which impairs pathogen's spreading and cellular recognition (208). Airway mucus involves 22 heavily glycosylated proteins that form high-molecular-weight mucus secretions (68,100,104,144). Although mucus secretion is considered a defensive innate tool, hypersecretion is a mark of disease progression. Mucus hypersecretion is not favored as it hampers gas exchange, obstructs airways, and results in breathing difficulty. This phenomenon is of great importance in infants where airways are already small and can be obstructed easily (116). HRSV infection induces mucus overproduction that forms clots with cellular debris causing airway inflammation and hyperresponsiveness (116,171,189).
Mucus secretion is controlled by immune receptors such as CXCR-2 (157), TLRs (TLR-3 and TLR-7) (211), and cytokines (IL-13, IL-17, and IL-23) (189,266). HRSV-infected TLR-3− mice showed Th-2 cytokines associated with IL-13, excessive mucus, and eosinophilia (211). Upregulation of IL-13 and IL-17 enhances mucus secretion in HRSV-infected mice. Inhibition of IL-17 through antibody neutralization dramatically reduced mucus secretion in mice infected with HRSV (163,244). Interestingly, there is differential expression of mucus among different viruses as well as among viruses of the same type. Differential expression of mucin in epithelial cell culture infected with HRSV and human metapneumovirus was observed (16). Similar results were also noticed in vitro, where the HRSV clinical isolate induced high levels of mucin compared with the A2 laboratory strain (258).
Pathogen recognition receptors
Recognition of invading pathogens through pathogen recognition receptors is an important event in initiating innate immune defense. Pathogen-associated molecular patterns can be recognized by carbohydrate-binding proteins (lectins), TLRs, and RIG-like receptors (RLRs). C-type lectins expressed by macrophages and DCs bring about pathogen ingestion and presentation. In addition, they trigger signal transduction and subsequent release of inflammatory cytokines (118). TLRs recognize and trigger an innate immune response against a wide array of pathogens, including bacteria, fungi, and viruses (5,7,205,222). TLRs expressed by sentinel cells such as lung resident macrophages, DCs, and lung epithelial cells recognize conserved HRSV motifs and launch an inflammatory response (11,211,219,239). HRSV infection upregulates expression of TLR-3, TLR-4, TLR-7, and TLR-8 during the first 24 h postinfection (63,88,161). TLRs activation mediates the production of potent antiviral cytokines IFN-α/β, which restrict virus replication in the early stages of infection (17,137).
HRSV surface glycoproteins, ss RNA, and ds RNA (replication intermediate) serve as ligands for TLRs (6,101,102). Interaction of HRSV ligands and TLR-2/TLR-6 was found to activate innate immune response by induction of inflammatory cytokines, migration of neutrophils, and DCs' activation in the lung (164). Signaling cascade following TLR-2/TLR-6 activation promotes the release of inflammatory cytokine IL-6 (149), chemokines (CCL-5, CCL-2) (63,178,243), and TNF-α early during HRSV infection (<24 h) (164). Some of these mediators augment lung pathology through necrotizing epithelial cells (TNF-α) (114), mobilizing more neutrophils, and induction of Th-2-biased immune response (CCL-2) (90). TLR-4, a surface TLR, interacts with the HRSV F protein by the aid of CD14 as a coreceptor (101,131). TLR-4-F interaction launches an innate immune response through the NF-κB pathway (93).
The importance of TLR-4 to HRSV infection was investigated in infants (112) and children, where TLR-4 mutations negatively affect NF-κB activation and subsequent events (252). Furthermore, preterm infants with TLR-4 signaling deficiency showed increased RSV pathology (12). Further studies demonstrated that inhibition of TLR-4/CD14-F complex during HRSV infection banned the release of IL-6 and IL-8 (176). Moreover, TLR-4 knockout mice displayed HRSV persistence, decreased mobilization of NK cells and CD14
HRSV infection upregulates expression of intracellular TLR-3, which in turn interacts with HRSV-ds RNA (2,88,133). Such interaction activates a signaling cascade that initiates the release of IFN-α/β, CXCL10, IL-8; CCL-12, CCL-5, and TNF-α (28,88,210). Dou et al. reported that increased expression of TLR-3 was associated with high levels of inflammatory cytokine TNF-α (63). TLR-3− mice infected with HRSV showed eosinophilia, Th-2 polarization, IL-13, IL-5, and mucus hypersecretion (211). Intracellular recognition of HRSV ssRNA by TLR-7 initiates the release of IL-12 and IL-23, which are required for T-cell differentiation (143). Other intracellular receptors that recognize ds RNA involve RIG-I and MDA5. These receptors belong to the RLR family, which is expressed by a variety of cells such as epithelial cells, macrophages, and conventional DCs.
Expression of RIG-I and MDA-5 is upregulated during HRSV infection with a subsequent release of IFN-β, CXCL10, and CCL-5 (146,217). Taken together, all the above data, incorporation of TLRs agonists as adjuvants should be considered in HRSV vaccine formulations. Johnson et al. recorded that administration of the TLR-9 agonist (CpG) but not TLR-7/8 agonists during the course of immunization markedly decreased disease severity following challenge with HRSV. However, administration of both TLR-7/8 and TLR-9 agonists during primary infection had a negative effect, where they enhanced airway inflammation (118). Thus, the timing and type of TLR agonists are critical factors in determining the disease outcome (118).
Neutrophils
HRSV interaction with epithelial cells drives the release of IL-8, the principle neutrophil chemoattractant (80). As they are the first to be recruited, neutrophils form the major participant of inflammatory response in the airway mucosa (69,255). HRSV-infected children exhibited neutrophilia in their bronchoalveolar lavage (BAL) (19). The percentage of neutrophils in HRSV-infected infants was estimated as 93% and 76% in the upper and lower parts of the respiratory tract, respectively (69). Neutrophil recruitment was observed during the first 48 h post-HRSV infection of mice (255). In vitro studies showed that the intercellular adhesion molecule (ICAM-1) establishes strong binding of neutrophils to HRSV-infected cells. Expression of ICAM-1 molecules is upregulated by IL-1 and CD18 integrin, which induced during HRSV infection (70,187,237).
Once interacted with HRSV-infected cells, neutrophils launch their cytotoxicity that kills infected epithelial cells (262). It was found that complement proteins secreted by epithelial cells enhance neutrophil killing function (271). In vitro interaction of HRSV with neutrophils leads to the release of IL-8, MIP-1α, MIP-1β, and granule-associated myeloperoxidase (MPO) (115). This finding was demonstrated in vivo by an increased level of MPO and neutrophil elastase in nasopharyngeal secretions of HRSV-infected children (1,79).
Activation of neutrophils causes the release of free oxygen radicals, elastase, and proteolytic enzymes (158). Another important defense mechanism launched by neutrophils is the formation of neutrophil extracellular traps (NET) that act to contain infection. In addition, chromatin fibers of NET are supported by neutrophil elastase and MPO (37). Such a complex was reported in HRSV infection and triggered by HRSV-F-TLR-4 interaction (75). In contrast, the excessive infiltration of neutrophils into lungs enhances disease progression. This was evidenced in infected infants, where increased neutrophil counts were associated with bronchiolitis (154). NETs formation was found to cause tissue damage in HRSV-infected children and enhance bronchiolitis. Coculture of neutrophils with HRSV-infected epithelial cells resulted in enhanced cell damage and detachment (262). Moreover, neutrophils secrete inflammatory cytokines (IL-8) and induce mucus production by excretion of TNF-α, which potentiates injury of healthy and infected tissues (Fig. 1) (19,40,119,261).
NK cells
Lungs contain the highest number of NK cells compared with other nonlymphoid tissues. NK cells account for 10% of the lung resident lymphocytes, which indicates their crucial role in lung protection against invading pathogens (227). Lacking NK cells due to genetic disorders or intended depletion renders humans and mice more vulnerable to respiratory infections (182,183,232). NK cells are among the early recruited leukocytes during HRSV infection. Mobilization of NK cells from peripheral circulation to the lungs was evidenced from the low blood counts in HRSV-infected children compared with the control group (57).
In mouse models, NK cells were detected on the 3rd day of HRSV infection (108,140). Impairment or depletion of NK cell migration in mice was associated with Th-2 immune response and eosinophilia (2,249). NK cells are important to initiate the Th-1 response (3,25,73). In agreement with this finding, Kaiko et al. demonstrated that NK cell depletion during HRSV infection induces polarization toward the Th-2 response through IL-25 released from epithelial cells (120). HRSV proteins G and SH were found to regulate the recruitment of NK cells to the lungs. HRSV mutants lacking G and SH are associated with increased levels of NK cells in BAL, suggesting that both proteins inhibit NK cell migration (57).
Several cytokines secreted by immune and nonimmune cells regulate NK cytotoxicity, proliferation, and migration. These cytokines include IL-12, IL-15, IL-18, IFN-γ-inducing factor, MIP-1α (CCL-3), MIP-1β (CCL-4), MCP-1, -2, -3, and CCL-5 (31,96,109). Of these, IL-15 and IL-12 are essential for NK cell development and their cytotoxic activity (30,124). MIP-1α regulates NK cell migration, which is demonstrated by both in vitro and in vivo studies (31,181). Mice lacking MIP-1α gene failed to induce a proper inflammatory response to HRSV (92). Moreover, IL-12 drives the release of IFN-γ from NK cells or CD8+ T cells, which inhibits the development of eosinophilia and enhances virus removal (108).
Cytokines released by activated NK cells have antiviral activities and they affect the proliferation and differentiation of other immune cells. Several studies demonstrated that NK cells release a number of cytokines such as IL-3, IL-8 (225), and IFN-γ (22,140). NK cell cytokines are also involved in differentiation of CD4+ and CD8+ T cells and maturation and activation of macrophages and DCs (14,128,207,259). In contrast, NK cells may exert their cytotoxic activity against macrophages and DCs (44,192).
The role of NK cells in HRSV disease progression was investigated. Upon activation, NK cells upregulated expression of NKG2D/CD27 receptors, which recognize HRSV-infected cells and launch their killing. Furthermore, they release high levels of IFN-γ, which cause cell damage (140). Interestingly, IFN-γ secreted by NK cells was found to inhibit the development of effective antibody responses to the HRSV in neonatal mice (247). Mice where NK cells were depleted displayed increased antibody titers (97,140,247), reduction in leukocyte extravasation, small amounts of IFN-γ, and substantial reduction in lung injury following HRSV infection (97,140).
Dendritic cells
DCs are among professional antigen-presenting cells (APCs) that mediate initiation and modulation of the adaptive immune response (145). There are several types of DCs that occupy different niches in the body (48). Thus, they act as sentinel cells that ingest and present pathogens to lymphoid cells. DCs have two different origins, myeloid and lymphoid. Myeloid-derived DCs (mDCs) reside in peripheral tissues as immature cells that are characterized by low expression of surface molecules such as CD80 (B7.1), CD86 (B7.2), and CD54 (ICAM-1) (54). The immature phenotype of mDCs, lung resident, was proposed to deliver the activation signals to naive T cells (220). This is due to the ability of immature DCs to ingest antigens either by phagocytosis or pinocytosis and migrate to lymph nodes or the spleen (89,134,220). However, naive T-cell activation and proliferation were completely diminished when mDCs got infected with HRSV (46,54).
Maturation of DCs is induced by several stimuli such as CD40 ligand (CD4+ T cells), GM-CSF, TNF-α and IL-1, bacterial-derived components (216,267), viral dsRNA (42), CpG motifs (256), or a monocyte-conditioned medium (26,41,215). In contrast, cytokines that are known to suppress immune response such as IL-10, TGF-β (8,54,177), and vascular endothelial growth factor (VEGF) (184) deactivate DCs maturation. Activated DCs upregulate the expression of NF-κB (86), present major histocompatibility complex (MHC) class II-antigen complex, and express costimulatory molecules required for T-cell activation and CD83 (274).
HRSV infection is associated with increased counts of mDCs and plasmacytoid dendritic cells (pDCs) in the lungs and surrounding draining lymph nodes (233). Lukens et al. reported excessive extravasation of mDCs (CD103
Lung mDCs induced the release of proinflamatory cytokines IL-17 and IFN-γ from naive T cells indicating their role in asthma. However, they were reported to secrete IL-12, a potent inducer of Th-1 cytokines (134). The type and strength of T-cell response due to interaction with mDCs are determined by a number of factors such as period of interaction, amount of presented antigen, costimulatory molecules, and released cytokines by DCs (223). pDCs are important in the early phase of HRSV infection, whereas mDCs expansion appears after the establishment of CTL response (160,223). The role of pDCs was proven in vivo where their depletion resulted in increased HRSV titers, mucus secretion, Th-2 cytokines, and exacerbated airway inflammation in mice (233) and led to development of bronchiolitis in children (265).
Although nonstructural proteins of HRSV (NS1 and NS2) are known to inhibit IFN-α/β production by many types of cells, including mDCs, pDCs release high levels upon HRSV infection, which potentiate their antiviral and antiallergic role (234,260). In vivo studies demonstrated that pDCs lasted throughout the course of infection and resolution (260). In a comparison between allergic and healthy children, pDCs counts were low in allergic ones, which highlighted their role in allergy alleviation (94). Consistent with this finding, children with pDCs deficiency developed asthma after severe HRSV-induced bronchiolitis (230). The contribution of mDCs in asthma development after HRSV bronchiolitis was reported. mDCs were able to sensitize mice to allergen and subsequent Th-2 responses (135). Moreover, in situ activation of naive T cells by DCs with Th-2 recall response potentiated the likelihood of allergen sensitization (52,55,254).
Macrophages
Macrophages are a member of APCs that have an instrumental role in modulation of the immune response. Activation of alveolar macrophages is mediated by IFN-α/β and IFN-γ released from infected lung cells. IFNs are also essential for proper antigen processing and presentation by macrophages (221,228). Alveolar macrophages have the ability to ingest and destroy all invading pathogens, including viruses (112,162). Once alveolar macrophages engulf an antigen, they release a number of cytokines (20,185,196). These cytokines regulate the inflammatory response and act to activate and recruit other lymphocytes into the lungs (155,173).
HRSV infection mobilizes more macrophages toward the lungs, where they play a defending role in case of acute infection (59,85). Activated macrophages release significant amounts of TNF-α, IL-6, CCL-3, IFN-α, CCL-5, and IFN-γ during early stages of HRSV infection (196). Interestingly, the efficiency of cytokine production especially IL-6 and TNF-α is less in neonates than in adults. This may explain in part the disease severity of RSV infection in infants (34,149). However, excessive secretion of proinflammatory cytokines may exacerbate airway inflammation (196). To investigate the role of macrophages during HRSV infection, macrophage depletion was attempted in several studies.
Intranasal inoculation of mice with clodronate, a macrophage cytotoxic molecule, resulted in deactivation of NK cells, impaired their migration into lungs, and increased virus replication (27,196). In such an environment, DCs emerge as the key effector cells of pulmonary innate response (112,246). Remarkably, alveolar macrophage depletion had no effect on initiation of adaptive immunity where CD4+ and CD8+ T cells were recruited and activated normally (196). In contrast, low number or depletion of alveolar macrophages enhanced lung immunopathology and increased viral load in HRSV-infected mice (204).
Although macrophages help to clear viral infections, in vitro and in vivo studies revealed that they provide a suitable site for viral replication and persistence (21,45,169,172,185). In vitro culture of alveolar macrophages was efficiently infected with HRSV. However, macrophages did not support a complete infectious cycle where only antigenic proteins but not complete virions were detected (203). Similarly, in vivo infection of lung macrophage was reported in individuals with acute HRSV infection (116). Moreover, phagocytosed HRSV can persist for a long time within macrophages (203). Such persistence is associated with intracellular modifications in gene expression, which affect macrophage functions. It was found that HRSV persistence enhanced phagocytosis through upregulation of FcγR (phagocytosis-mediated receptor), IL-1, and IL-6 (32,76,175). Moreover, HRSV persistence in alveolar macrophages resulted in chronic inflammation of the lower respiratory tract (36).
Viruses impair macrophage activation through inhibition of IFNs induction through several mechanisms. These mechanisms include deactivation of STAT (84), intracellular degradation of STAT proteins (67,202), sequestration of STAT proteins in high-molecular-weight complexes (206), and inhibition of nuclear localization of STAT proteins (241). Proteasomal degradation of STAT2 is the mechanism adopted by HRSV to inhibit type I IFN signaling by nonstructural proteins NS1 and NS2 (201,202) or by inhibition of tyrosine kinase-2 phosphorylation (226).
Eosinophils
Eosinophils have long been known as the principle innate immune cells to fight helminthic pathogens. They are members of granulocytes, which are characterized by having cationic granule proteins (105,132,251), reactive oxygen species (213), lipid mediators (15), and growth factors (TGF, VEGF) (107,123). Eosinophils also secrete a wide array of cytokines: IL-1, IL-2, IL-4, IL-5, IL-6, IL-8, IL-13, and TNF-α (105). Moreover, eosinophil granules possess ribonucleic acid degrading enzymes (RNases) that enforce their role in fighting viral infections (61). This was testified in vitro where eosinophils' capacity to restrict virus replication was hampered in the presence of a ribonuclease inhibitor (62,209).
Eosinophils can also recognize viral genome through several TLRs such as TLR-1, TLR-4, TLR-7, TLR-9, and TLR-10 through which they can initiate innate and acquired immune responses (168,191). HRSV ssRNA acts as a ligand for eosinophilic TLR-7, which upon activation can stimulate degranulation through the MyD88-dependent mechanism (191). Phipps et al. also observed that increased counts of eosinophils enhanced virus clearance from lungs of HRSV-infected mice and prevented airway inflammation (191).
HRSV infection initiates mobilization of eosinophils from peripheral blood into human lungs (69,99,275). In vitro studies revealed that eosinophil degranulation is efficiently induced by HRSV-infected epithelial cells (181). Epithelial cells respond to HRSV infection by releasing a number of cytokines, chemokines, and growth factors that control eosinophil mobilization and degranulation (Fig. 1) (22,150). These mediators include TNF-α (187), GM-CSF, RANTES (22,181,214), and MIP-1α (78). Furthermore, in vitro culture of HRSV-infected epithelial cells increases expression of surface molecules such as ICAM-1 and CD18 (237), which facilitates eosinophil binding and subsequent diapedesis (33). HRSV infection of eosinophils was investigated in vitro and confirmed by immunofluorescence (214) and transmission electron microscopy (126). Such infection triggers a signaling cascade that drives eosinophils to secrete oxygen radicals, hyperoxide, leukotriene C4, RANTES, and MIP-1α (240). RANTES and MIP-1α mediate activation of eosinophils in an autocrine manner (180).
The role of eosinophils in the development of lung inflammation was clearly investigated in both in vivo and in vitro studies. Eosinophilia was a characteristic marker in dead infants who received formalin-inactivated RSV vaccine (24,47,235,244). Immunohistological analysis revealed the deposition of eosinophils and their granule contents on the damaged airway epithelia of asthmatic patients (35,98). Similarly, in in vitro assays, eosinophils exerted the same effect against epithelial cells derived from the respiratory tract (13). The most prominent eosinophilic toxic protein detected in damaged tissues is eosinophilic cationic protein (ECP).
Levels of ECP were higher in nasopharyngeal secretions from children with HRSV-induced bronchiolitis, suggesting its contribution to airway obstruction (229). This finding was observed in vitro where HRSV-infected epithelia supplied with eosinophils secreted large amounts of ECP through the CD18-dependent mechanism (181). Eosinophilia can be induced indirectly by the HRSV attachment G protein. G protein has long been known to induce Th-2 cytokines, which in turn enhance eosinophil movement into lungs (9,10,236). This feature is linked mainly to the secreted form of G protein. Mice immunized with vaccinia virus vector expressing soluble G protein (i.e., not membrane anchored) displayed increased IL-5 production; mobilization of eosinophils increased illness and decreased viral expulsion (117).
The role of IL-5 in exacerbation of airway inflammation was also investigated in IL-4, IFN-γ, and IL-5-deficient mice. Eosinophilia and subsequent lung inflammation were recorded in mice lacking IL-4 and IFN-γ following HRSV challenge. In case of IL-5 deficiency, mice showed no airway inflammation. Moreover, airway inflammation was restored by injection of IL-5, which confirms its role in mobilization of eosinophils to the lungs (224). Although there is a body of evidence supporting the contribution of eosinophils in airway inflammation, other studies indicated a role for other leukocytes.
Neutrophils were the predominant cells associated with vaccine-enhanced RSV lung inflammation in African green monkeys and calves (82,121). Similarly, Everard et al. observed an increased number of neutrophils in BAL fluid from infants with HRSV bronchiolitis (69). In conclusion, eosinophils play an important role during HRSV infection, where they can act as APC (264). In contrast, they may contribute to obstruction of airways by killing of HRSV-infected alveolar epithelial cells as well as secreting inflammatory mediators.
HRSV Vaccine and Immune Response
A good vaccine should evoke specific immune response without potentiating disease progression. Unfortunately, attempts to produce a fully protective HRSV vaccine were not successful. The first vaccine was a formalin-treated RSV, which unluckily led to disease enhancement and death of two vaccinees (47). Understanding the immunopathology associated with formalin-inactivated (FI)-RSV is required for developing safe and effective vaccines. Data obtained from children and animal models showed that immunological changes due to FI-RSV were due to a Th-2-biased immune response associated with eosinophilia (24,47,56,117,125,160,235,242,263).
In addition, decreased levels of neutralizing antibodies (167) and deposition of immune complexes (antigen antibody and antigen-complement complexes) were detected in FI-RSV-treated mice (193). The same result was monitored in a lung biopsy derived from children with improved RSV immunopathology (193). Another possible explanation is that formalin induces conformational changes to HRSV antigenic proteins. Such changes weakened the fusion protein signaling through TLR-4, which impairs subsequent events (58).
To avoid the adverse effects of FI-RSV, a number of vaccine formulations were described, including live attenuated, subunit, virus-like particle (VLP), and gene-based vaccines. Live-attenuated vaccines are produced either by traditional methods (cold passaged/temperature sensitive) (74,153,197) or by reverse genetic techniques (Fig. 2) (51,148). Live-attenuated vaccines were proven not to enhance disease upon challenge with wild-type viruses, besides they were capable of inducing innate, cellular, and humoral immunity (51,122,268).
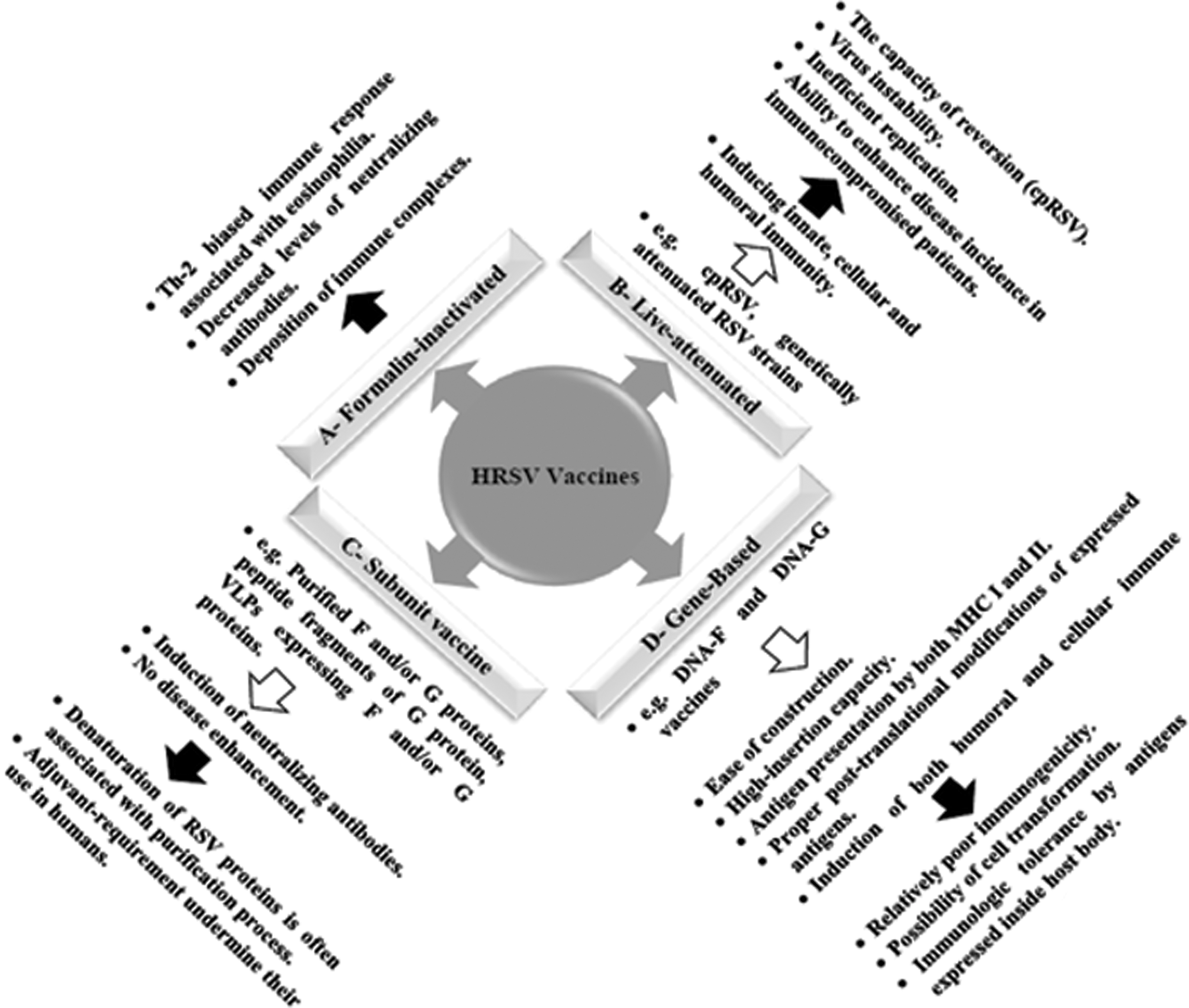
A diagrammatic representation of different HRSV vaccine categories. White arrows refer to advantages, while black arrows refer to shortcomings of each category.
In a phase I clinical trial, children immunized with live-attenuated RSV vaccine had low levels of IFN-γ, IL-1β, IL-2, IL-6, and IL-13. This study also demonstrated an inverse correlation between the degree of attenuation and cytokine release (122). However, a number of limitations were reported, such as (i) the capacity of reversion, (ii) virus instability that hinders vaccine production and storage (122), (iii) inefficient replication of attenuated virus does not evoke effective immune response (269), and (iv) ability to enhance disease incidence in immunocompromised patients (121a).
Several subunit vaccine formulations were developed to overcome the limitations of live-attenuated vaccines. These formulations include (i) purified F and/or G proteins (71,72,82,136), (ii) peptide fragments of G protein (194), and (iii) VLPs containing F and/or G proteins (Fig. 2) (152,198). Purified F proteins, designated PFP-1, PFP-2, and PFP-3, were capable to induce high titers of neutralizing antibodies in clinical trials (23,87,186,250).
A protein mixture of G, F, and M proteins was tested in cotton rats and in clinical trials (phases I and II), inducing a powerful level of neutralizing antibodies with no evidence of disease enhancement upon virus challenge. However, there are no data about the protection efficacy of such a mixture (166). The use of BBG2Na, a synthetic peptide spanning the amino acid residues 130–230 of G protein and combined with Streptococcus G protein, was able to protect cotton rats from lower RTIs (195). The potential denaturation of BBG2Na during protein purification and the need of adjuvant undermined the use of such a vaccine candidate in humans (165). VLPs expressing G and/or F proteins without adjuvant induced high antibody titers in mice (151,164).
The progressive advance in the field of recombinant DNA technology is continuously providing the tools for generating novel promising vaccine candidates against HRSV. Immunization with the DNA vaccine mimics the action of live attenuated viruses where the target gene is expressed intracellularly and presented in conjunction with both class I and II MHC molecules. DNA vaccines are suitable for newborns since the transgene cannot be recognized by preexisting maternal antibodies (91). Several attempts were carried out to enhance the immunogenicity of DNA vaccine candidates. These enhancers include CpG DNA motifs, bacterial cell wall components, or molecular adjuvants such as CD40L (129,248). Others reported that incorporation of DNA plasmids encoding cytokines helps to modify the immune response (24a).
The efficacy of DNA plasmids expressing HRSV F and/or G proteins as potential vaccine candidates was investigated. DNA-F plasmids were reported to mimic the immune response of natural virus infection in mice by inducing neutralizing antibodies, CD8+ T cells, and IFN-γ, besides conferring protection against the challenge virus. Moreover, the DNA-F plasmid can establish an efficient Th-1 response associated with protective immunity and switch preestablished Th-2 response (responsible for enhanced disease progression) to Th-1 (142,179,190). Proper expression of DNA plasmids encoding F protein requires codon optimization to avoid premature polyadenylation caused by cryptic polyadenylation signals and splicing sites (275).
Although G protein is less conserved among RSV subgroups, DNA-G plasmids that encode membrane-anchored or secreted forms were capable to induce balanced systemic and pulmonary Th-1/Th-2 responses in mice with no signs for disease enhancement upon challenge. However, G-DNA vaccines were unable to induce virus-specific cytotoxic T-cell responses (141). Vaccine formulations such as live attenuated and recombinant DNA vaccines require adjuvants to enhance their immunogenicity. Monophosphoryl lipid and protollin initiate TLR-4 signaling cascade, which suppresses polarization of naive CD4+ T cells into Th-2 cells (50). Incorporation of DNA segments that code for cytokines enhances immune response of recombinant DNA-based vaccines. A number of cytokines that have a regulatory immune effect were used such as IL-12, GM-CSF (38,60,64,111), IFN-γ, IL-27, and IL-23 (39,272).
Conclusion
Although HRSV is known to cause substantial epidemics worldwide, trials to produce a vaccine were not fruitful. This could be attributed to the virus ability to circumvent the immune system. HRSV interaction with lung epithelial cells initiates a cascade of events, which involves the recruitment, activation, and proliferation of leukocyte population (neutrophils, NK cells, DCs, macrophages, and eosinophils). These cells release a pool of cytokines, chemokines, and other mediators that orchestrate and potentiate the immune response. The main goal of these mediators is to contain infection and remove viral particles. However, during HRSV infection, there is a massive infiltration of leukocytes associated with overproduction of immune mediators. Hypersecretion of these mediators (such as TNF-α, ECP, NETs, IFN-γ, and IL-8) leads to tissue damage and subsequent deterioration to bronchiolitis, pneumonia, and even asthma. Therapeutic agents that enhance innate immune response may help to alleviate disease outcome and restrict virus propagation. Overall, a balance between innate immune elements should exist to avoid the harmful side of the innate immune response. There is a large body of research regarding improvement of the innate immune response against HRSV. Of these, genetic-based vaccines, drugs targeting HRSV receptors, and TLR activation are promising to generate more effective therapies.
Footnotes
Acknowledgment
This work was partially supported by NSTIP strategic technologies program number (14-MED809-02), King Saud University, Kingdom of Saudi Arabia.
Authors' Contributions
Mohamed A. Farrag and Fahad N. Almajhdi cowrote the article, checked for the literature, and adapted the bibliography. All authors read and approved the article.
Author Disclosure Statement
The authors declare that they have no competing interests.