
Abstract
Hepatitis E virus (HEV) is one of the primary causative agents of acute hepatitis. It is noteworthy that HEV can develop chronic infection and even lead to liver cirrhosis; however, the mechanism has not been revealed. In this study, the ELISA assay was used to detect protein levels, and we found that HEV open reading frame 3 (ORF3) protein inhibited the expression of proinflammatory cytokines (tumor necrosis factor-alpha [TNF-α], interleukin [IL]-1β, IL-6, IL-8, IL-12p40, and IL-18) and chemotactic factors (nitric oxide [NO], interferon-inducible protein-10 (IP-10), macrophage inflammatory protein (MIP)-1α, monocyte chemoattractant protein-1 (MCP-1), granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF)] in lipopolysaccharide (LPS)-stimulated human PMA-THP1 cells. Further study showed that mRNA and protein levels of pattern recognition receptors (PRRs), such as Toll-like receptor 4 (TLR4), TNF receptor-associated factor 6 (TRAF6), and nucleotide-binding oligomerization domain containing 2 (NOD2), decreased after infection of pLL3.7-ORF3 (pORF3); moreover, the inhibition produced corresponding upregulation of IκBα and downregulation of phosphorylated IκB kinase IKKɛ (p-IKKɛ) and phosphorylated nuclear factor (NF)-κB (p-NF-κB), but little variation was found in the concentration of IKKɛ and NF-κB. Taken together, our results demonstrated that HEV ORF3 attenuated LPS-induced cytokine production and chemotactic factors, predominantly by inhibiting various PRRs-mediated NF-κB signaling pathways. The anti-inflammatory properties might be of great importance to clarify the role and mechanism of macrophages in chronic HEV infection and cirrhosis.
Introduction
H
The HEV genome is a single-stranded RNA of ∼7.2 kb that is positive sense, with a 5′-methylguanine cap and a 3′ poly (A) stretch. It contains three partially overlapping open reading frames (ORFs)—designated ORF1, ORF2, and ORF3. ORF1 (3) encodes a nonstructural protein, ORF2 encodes the capsid protein, and ORF3 encodes a protein of only 114 amino acids (13) that contains two hydrophobic domains (D1, D2) at the N-terminus and two proline-rich domains (P1, P2) at the C-terminus (17).
In prior studies, two broad roles were predicted for the ORF3 protein in HEV pathogenesis. First, it promotes cell survival and proliferation through extracellular regulated kinase (ERK) activation, extended endomembrane signaling from activated growth factor receptors, and attenuation of the intrinsic death pathway. Second, ORF3 protein also dampens innate host responses through an attenuated acute phase response and increased secretion of immunosuppressive factors, such as alpha-1-microglobulin, or regulated the expression of proinflammatory cytokines (6, 9). Accordingly, ORF3 might be a viral accessory protein that regulated the host response toward infection. However, little is known about the detailed regulation mechanism of HEV ORF3 in macrophages. Therefore, we attempted to explore the underlying molecular mechanism for the inhibitory effects of HEV ORF3 on lipopolysaccharide (LPS)-induced cytokine production.
Natural immunity recognizes microorganisms through pattern recognition receptors (PRRs), which are described below. First, Toll-like receptors (TLRs), as representative PRRs, is an integral component of the inflammation process. It has been reported that TLR4 is the signal-transducing receptor for LPS (32), which can be second-strike for hepatargia (23, 25). Second, nucleotide-binding oligomerization domain containing 1 (NOD1) and nucleotide-binding oligomerization domain containing 2 (NOD2) are involved primarily in mediating antibacterial defenses (41). NOD1 and NOD2 can be associated with the bacterial adaptor molecule to activate nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) through CARD–CARD interaction, followed by induction of numerous genes involved in the inflammatory process (19, 20). Third, tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) functioned as an adaptor protein for various receptors, such as interleukin (IL)-1/Toll like, TNF-related activation-induced cytokine (TRANCE), and CD40-receptor, leading to NF-κB activation (22).
Previous studies have reported that NF-κB belongs to a downstream signaling component of TLRs, which has a feedback regulation on the transcription of PRR (22). NF-κB signaling pathway is a core part of regulation of a cellular inflammatory response (2), and p65 is a major subunit of the NF-κB signal transduction pathway. It is worth noting that HEV ORF3 can inhibit p65 in the liver cell translocating from the cytoplasm to nucleus (38), subsequently upregulating various proinflammatory factors playing a role in immunity.
Based on the previous research, macrophage has a key regulating role in the progress and ease of liver cirrhosis; therefore, what is the underlying mechanism? Immune cells can recognize pathogen-associated molecules, including LPS (4), which is derived from the outer membrane of gram-negative bacteria, and mannans of yeast cells through TLRs expressed on the cell surface. Humans have various PRRs, including TLRs, TRAF6, NOD2 (40), and NOD-like receptors (21). These receptors transduce signals to activate the NF-κB or MAPK pathway, which subsequently drives the induction of several proinflammatory cytokines and chemokines. The inflammatory response may play an important role in the HEV invasion (5, 28).
Previous studies suggest that STP-A11, a tumor protein, upregulates both the NF-κB and AP-1 transcription activity through TRAF6, which would ultimately contribute to cellular transformation (22); a similar role has been found in STP-C, another protein of herpesvirus saimiri (8). While another research demonstrates that protein HBV polymerase has preferentially suppressed the NF-κB pathway through inhibiting the activity of IKKs through interaction with Hsp90β (24). Accordingly, regulation of proteins exhibit difference on NF-κB signaling pathway. In this study, we attempted to investigate how HEV ORF3 affected the function of macrophages, and whether the effects are mediated by NF-κB pathways through PRR, which may clarify mechanisms of ORF3-involved inflammatory response.
Materials and Methods
Reagents and antibodies
Escherichia coli 0111:B4 LPS (Invitrogen, Carlsbad, CA), penicillin/streptomycin, and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum (FBS; Gibco BRL, Grand Island, NY) and human monocytic leukemia cell line THP-1 were purchased from American Type Culture Collection (ATCC, Manassas, VA). Plasmid pLL3.7, Δ8.9, and VSVG were bought from Thermo Fisher (Thermo, Beijing, China). All the primary antibodies were from Cell Signaling Technology (Danvers, MA) and Santa Cruz Biotechnology (Santa Cruz, CA), and HRP-conjugated secondary antibody was purchased from Promega (Madison, WI). All primers were designed and synthesized in Sangon Biotech (Shanghai, China).
Cell culture, infection, and treatment
THP-1 monocyte cell line was cultured in RPMI 1640 medium supplemented with 10% FBS, penicillin (100 μg/mL), and streptomycin (100 μg/mL). Cells were incubated at 37°C in 5% CO2 and then treated with 50 ng/mL of PMA for 48 h to induce differentiation into macrophages, and washed twice with phosphate-buffered saline (PBS). All the experiments were performed with THP-1-X Blue cells. After culturing for 3 h at 37°C in 5% CO2 atmosphere, cells were infected using the lentivirus packaged by pLL3.7 or pLL3.7-ORF3, Δ8.9 and VSVG in 293 T cells. Various lentiviruses were added in the presence of 8 ug/mL polybrene (Sigma-Aldrich, St. Louis, USA) including pLL3.7 empty vector and pLL3.7-ORF3 (pORF3) to PMA-THP1 cells, after mixed lightly, then cultured for 24 h. After 48 h post-infection, they were treated with 1 μg/mL LPS at 18 h or without LPS. RNA or protein was extracted as needed.
ELISA assay
The level of each proinflammatory cytokine and chemotactic factors in the cell culture supernatants was determined by the ELISA Kit (Shanghai Enzyme-linked Biotechnology Co., Ltd., Shanghai, China) according to the manufacturer's instructions. All samples were assayed in triplicate.
Real-time quantitative RT-PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA). The cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster city, CA, USA). PMA-THP1 cells infected with pLL3.7 and pORF3 were stimulated with or without 1 μg/mL LPS for 18 h. The expression of intracellular PRR (TLR4, TRAF6, and NOD2) mRNA was measured by real-time quantitative polymerase chain reaction (RT-qPCR) System (Applied Biosystems) and was standardized to β-actin mRNA. The mRNA levels of TLR4, TRAF6, and NOD2 were calculated with the ΔΔCt-method, and relative expression was expressed as fold expression over average of control cells.
Western blot analysis
Western blot was performed as described and THP-1 cells were stimulated with 1 μg/mL LPS or unstimulated after infection with pLL3.7 and pORF3. The cells were washed with cold PBS and then lysed in 150 mL lysis solution. The protein concentration was estimated using BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL). Equivalent amounts of protein were loaded onto 10% SDS-PAGE and electrophoretically transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat milk for 2 h at room temperature and then incubated at 4°C overnight with primary antibodies of TLR4, TRAF6, NOD2, IκBα, IκB kinase IKKɛ/IKK-i, phosphorylated IKKɛ (p-IKKɛ), NF-κB, and phosphorylated NF-κB (p-NF-κB) in a 10-fold diluted blocking buffer. The blots were washed three times with TBS and then incubated for 2 h using HRP-conjugated secondary antibody at room temperature. Immunoreactive bands were developed using an Amersham ECL Plus Western Blotting Detection System (GE Healthcare, Buckinghamshire, United Kingdom) and visualized by Molecular Imager VersaDoc MP Imaging Systems (Bio-Rad Laboratories, Hercules, CA).
Statistical analysis
Data analysis was performed using the SAS system (ver.9.1.3, SAS Institute Inc., Cary, NC) and presented as mean ± standard deviation values. Data for western blot analysis were presented as fold change over the respective control that was arbitrarily defined. Two-tailed unpaired Student's t-test was used. Differences with p < 0.05 were considered to be statistically significant.
Results
Inhibition of HEV ORF3 on LPS-induced production of proinflammatory cytokine in PMA-THP1 cells
In our study, we comprehensively evaluated the effect of ORF3 on various inflammatory cytokines. Initially, THP-1 cells were differentiated into macrophage cells by incubation with 50 ng/mL PMA for 48 h, then PMA-THP1 cells were infected with pORF3 using pLL3.7 plasmid as control, and finally, the level of proinflammatory cytokines were measured with the ELISA Kit after stimulating with 1 μg/mL LPS for 18 h. Test results indicated that LPS stimulation significantly activated the expression of cytokines, whereas cells infected with pORF3 showed obvious reduction of LPS-induced proinflammatory cytokines, including TNF-α (Fig. 1A), IL-1β (Fig. 1B), IL-6 (Fig. 1C), IL-8 (Fig. 1D), IL-12p40 (Fig. 1E), and IL-18 (Fig. 1F) when compared with the group infected with pLL3.7.
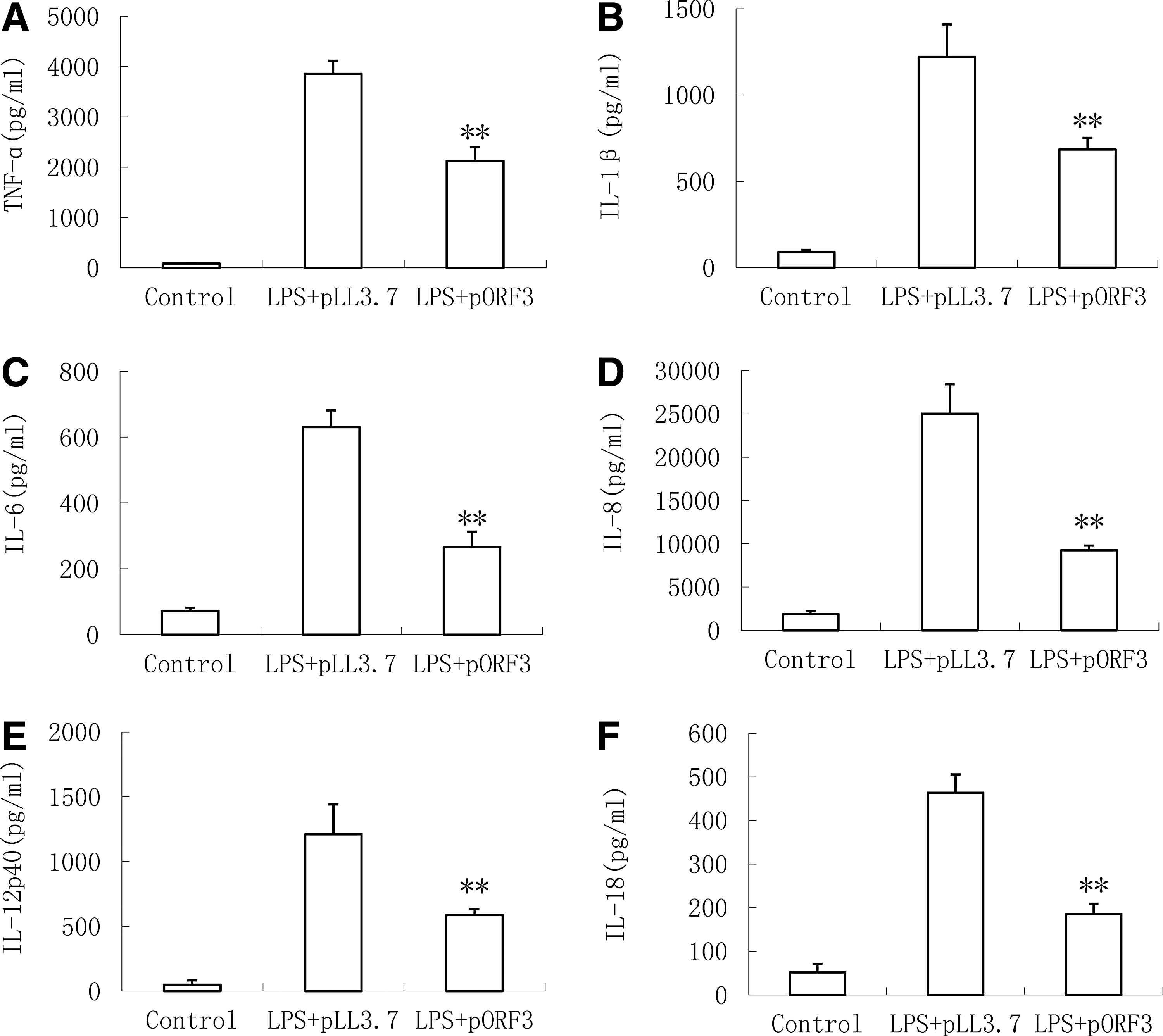
Inhibition of open reading frame 3 (ORF3) on lipopolysaccharide (LPS)-induced production of proinflammatory cytokines in PMA-THP1 cells. The concentrations of cytokines—
Suppression of HEV ORF3 on LPS-induced production of chemotactic factor in PMA-THP1 cells
To further investigate the impact of ORF3 on the function of macrophage, protein levels of various proinflammatory factors were detected by the ELISA Kit completely. As shown in Figure 2, in the presence of pLL3.7, LPS stimulation enhanced concentrations of chemotactic factors, including nitric oxide (NO, Fig. 2A), interferon-inducible protein-10 (IP-10, Fig. 2B), macrophage inflammatory protein (MIP)-1α (MIP-1α, Fig. 2C), monocyte chemoattractant protein-1 (MCP-1, Fig. 2D), granulocyte colony-stimulating factor (G-CSF, Fig. 2E), and granulocyte macrophage colony-stimulating factor (GM-CSF, Fig. 2F) compared with the absence of LPS (control) (p < 0.05). However, there is a measurable reduction in the level of IP-10, MIP-1α, MCP-1, G-CSF, and GM-CSF transiently infected with pORF3 cells stimulated with LPS.
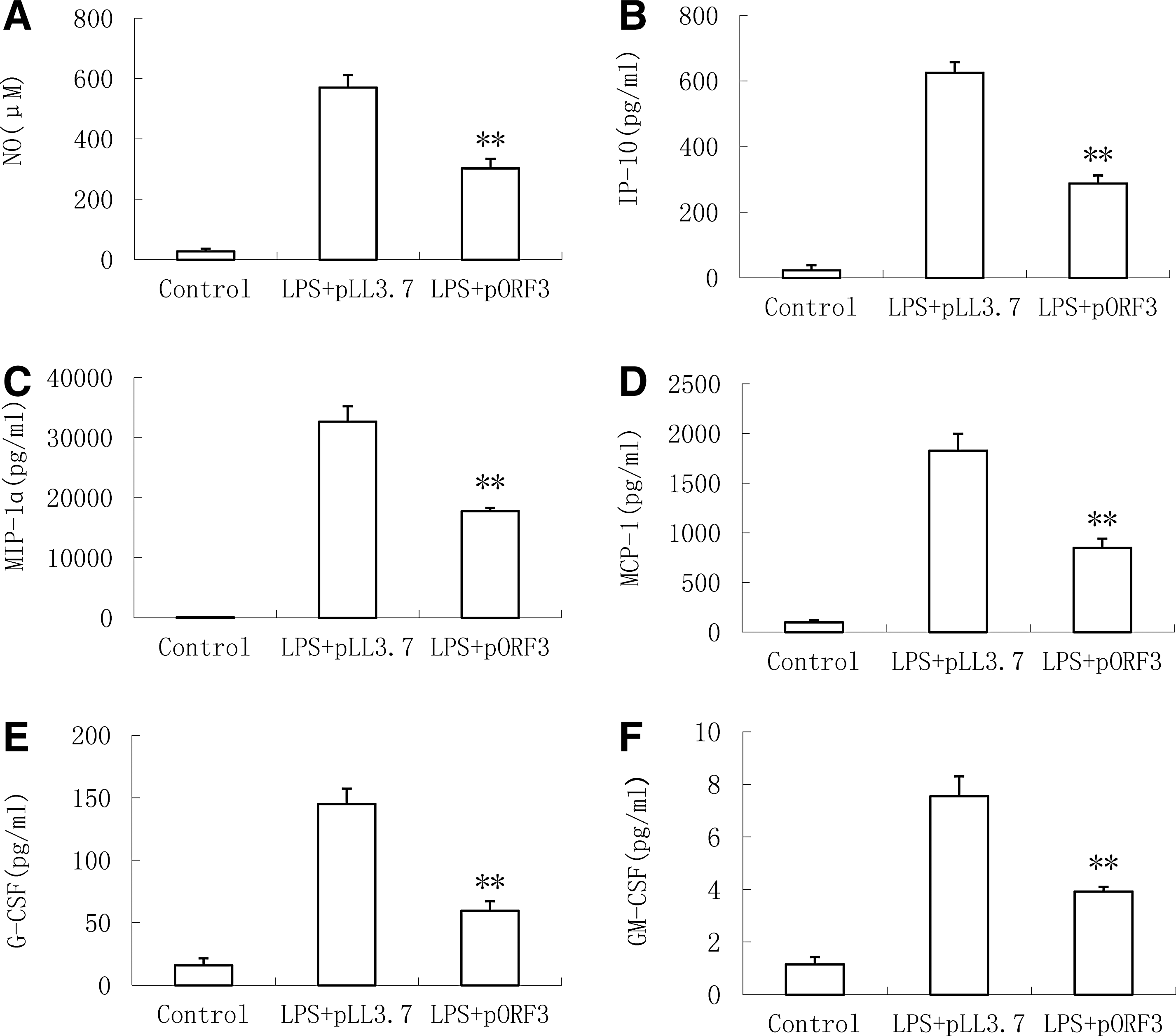
Suppression of ORF3 on chemotactic factors in PMA-THP1 cells stimulated with LPS. The concentrations of chemotactic factors—
HEV ORF3 suppressed LPS-induced expression of TLR4 TRAF6 NOD2 genes and protein concentration in PMA-treated THP-1 cells
To reveal the reactivity mechanism of LPS in PMA-THP1 cells following pORF3 infection, we observed transcription and translation of PRRs, such as TLR4, TRAF6, and NOD2, expressed in macrophage using RT-qPCR and western blot. The result showed that TLR4, TRAF6, and NOD2 mRNA significantly diminished in pORF3-infected cells compared with the group infected with pLL3.7 (p > 0.05) (Fig. 3A). Similar phenomenon has been found on the concentration of protein of these three genes using the ELISA Kit (Fig. 3B).
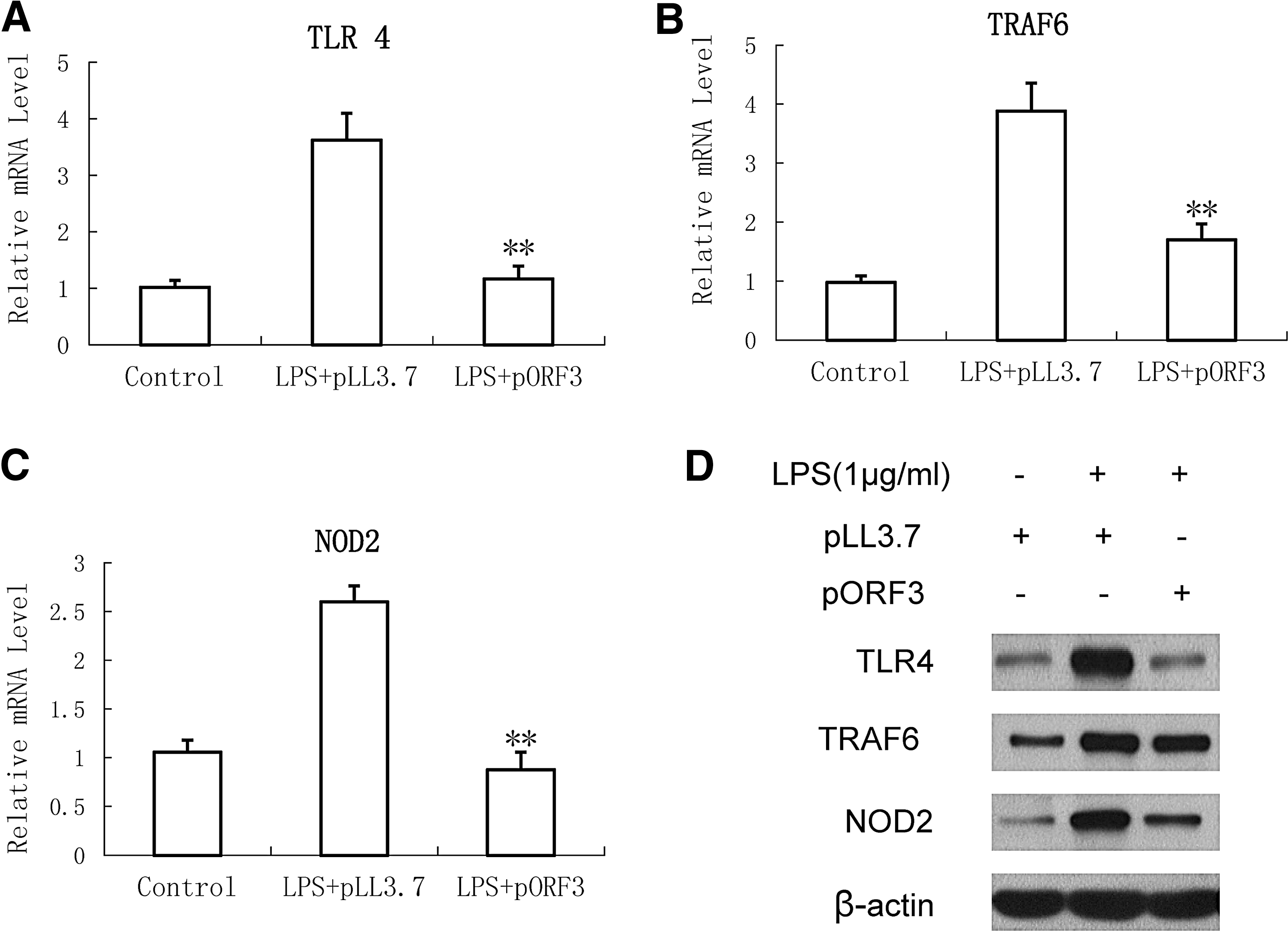
Inhibition of hepatitis E virus (HEV) ORF3 on protein level and gene expression of intracellular pattern recognition receptors in PMA-treated THP-1 cells. THP-1 cells were stimulated with 1 μg/mL LPS and cultured for 18 h or not after infection with or without pORF3. Expression of mRNA for Toll-like receptor 4 (TLR4)
NF-κB pathways inhibited by HEV ORF3 treatment in PMA-treated THP-1 cells stimulated with LPS
To persistently infect the host, many viruses have evolved strategies to evade antiviral immune defenses. Accordingly, to determine whether HEV ORF3 suppressed the level of proinflammatory cytokines and chemotactic factors through NF-κB pathways, western blot was used to measure the concentrations of IKKɛ, p-IKKɛ, IκBα, NF-κB, and p-NF-κB after LPS stimulation. As shown in Figure 4, compared with the control group, ORF3 obviously enhanced the level of IκBα but decreased the concentrations of p-IKKɛ and p-NF-κB, and it had no effect on the protein expression level of IKKɛ and NF-κB.
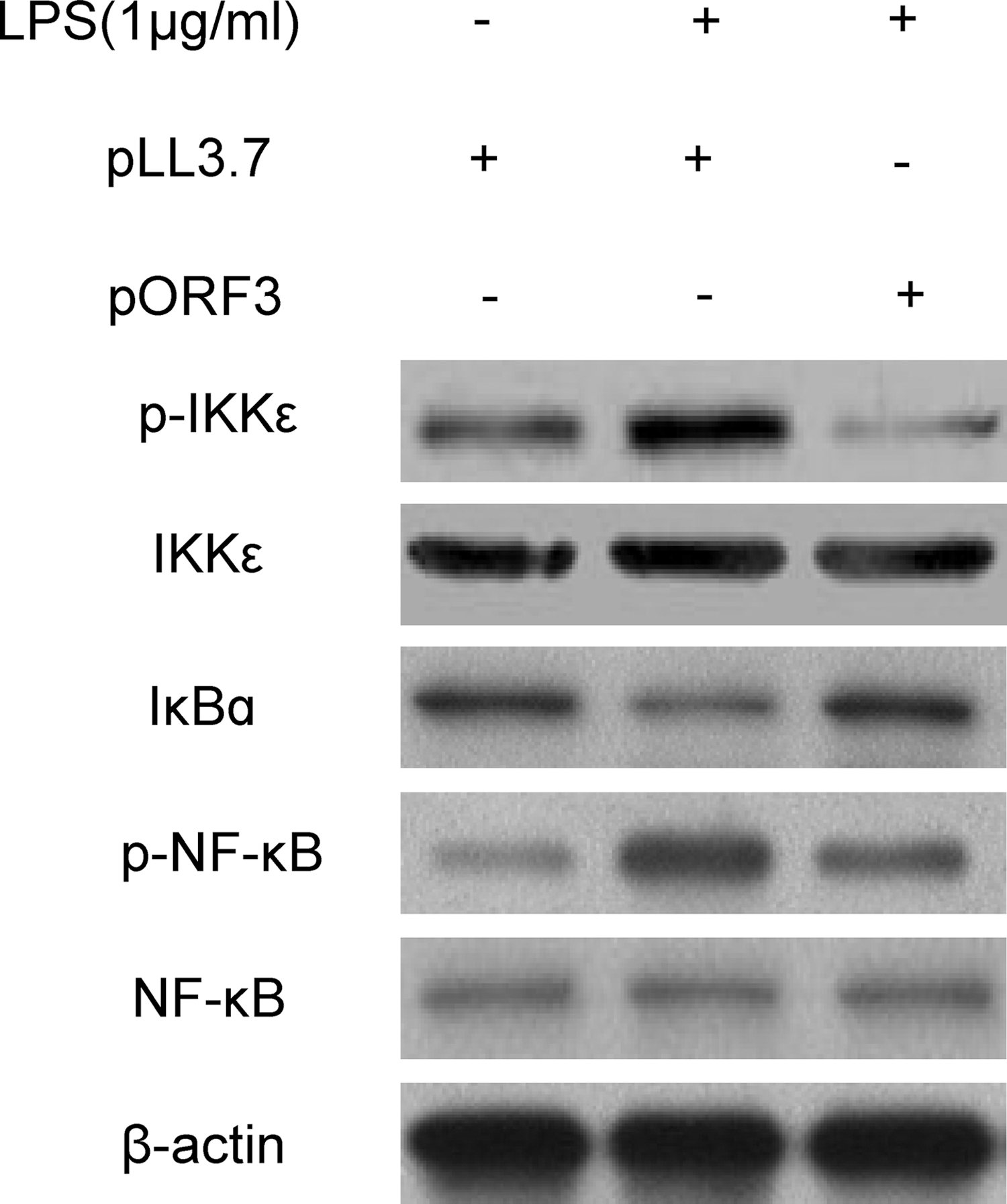
Effect of HEV ORF3 on LPS-induced activation of IκBα and phosphorylation of IKKɛ, nuclear factor (NF)-κB in PMA-treated THP-1 cells. The cells were infected with pLL3.7 and pORF3 and then left untreated or stimulated with 1 μg/mL LPS for 18 h. Western blot was used to analyze the levels of IKKɛ, p-IKKɛ, IκBα, NF-κB, and p-NF-κB.
Discussion
Macrophages, as an important component of the innate immune system, can secrete several inflammatory cytokines to eradicate pathogens following LPS stimulation, which play an important role in the control of virus infection. Besifloxacin (42), intravenous immunoglobulin (31), and Porphyromonas gingivalis (7), could adjust the level of LPS-induced inflammatory cytokines or chemotactic factors in THP-1 cells. As a pathogen, the HEV genome encodes the ORF1, ORF2, and ORF3 proteins, among which the ORF3 protein appears to have regulatory properties (29). Some literatures show that ORF3 attenuated the mRNA and protein expression levels of IL-1β, COX-2, ICAM-1 (39), and IFN-α (9) contributing to the innate immune response. HEV ORF3 protein also protects cells from mitochondrial depolarization and death. Although several studies of HEV ORF3 and its function on macrophages have been carried out recently, comprehensive analysis and study still have not been given. Consequently, our overall research analyzed the effect of ORF3 on various inflammatory cytokines or chemotactic factors, and results suggested that ORF3 significantly inhibited LPS-induced production of inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-8, IL-12p40, and IL-18). Inflammatory cytokines and chemokines play key roles in inflammation during a viral infection. For instance, IL-12 played a critical role in the early inflammatory response to infection and also in the generation of Th-1 cells, which favored cell-mediated immunity (18). Another research has found that HBeAg suppresses the production of IL-12/IL-6 from dendritic cells (DCs), which may contribute to the immune escape of HBV that supports persistent infection (35). Thus, we speculated that the HEV ORF3 suppression of inflammatory cytokines was able to modulate the host–cell environment, which might attenuate antiviral effects of host cells and contribute to efficient viral replication.
To further explore HEV ORF3 function in macrophages, we measured the concentrations of six chemotactic factors, including NO, IP-10, MIP-1α, MCP-1, G-CSF, and GM-CSF, and results showed that the concentrations diminished in pORF3-infected cells, but not in pLL3.7-infected PMA-THP1 cells after stimulation of LPS. The inhibitory effect is just another indicator of anti-inflammatory effect of HEV ORF3. Chemokines are a family of low-molecular-weight protein, and they can influence the outcome of infection by regulating the recruitment of inflammatory cells into tissues, which lead to the pathogenesis of hepatocellular carcinoma, adjusting the inflammatory microenvironment of the tumor, with evasion of the immune response (10, 26, 36). Therefore, the obvious attenuation of six chemotactic factors might suppress inflammatory response, thus causing the evasion of HEV from the immune response, and enabling it to establish chronic infections and associated liver disease.
LPS is a well-characterized inducer of inflammatory response and the proinflammatory activity of LPS-induced stimulation can be recognized by PRRs, therefore, we characterized the level of transcription and translation for three PRRs, TLR4, TRAF6, and NOD2, and a significant decrease was found in PMA-THP1 cells transferred with ORF3. In Waggoner's study, HCV could bind with core protein gC1qR through the PI3K pathway correlating with a failure in transcription of the IL-12p40 subunit mediated by TLR4, which contributed to the escape from immune and long-term survival of HCV (37). Based on the above results, we could speculate that HEV ORF3 suppresses recognition for TLR4, TRAF6, NOD2 and may induce changes in the downstream signaling pathway and lead to chronic infection.
Although HEV ORF3 inhibited the level of inflammatory cytokines and chemotactic factors, it weakened the recognition for PRRs, and it is unclear at present whether these effects are mediated by NF-κB or other transcription factors. Interestingly, previous research showed that ORF3 reduced the mRNA and protein expression levels of IL-1β, COX-2, and ICAM-1 through suppressing TNF-α-induced NF-κB activation (39). Moreover, another study reported that genotype 1 HEV ORF3 may transiently activate NF-κB through unfolded protein response in the early stage and subsequently suppress the NF-κB signaling induced by TNF-α in the late phase to create a favorable virus replicable environment (39). In addition, ORF3 can regulate other multiple signaling pathways, including those involving MAPK-c-Jun N-terminal kinase (MAPK–JNK1/2) and ERK. ORF3 activates the ERK by its binding and inhibition of Pyst1 (33).
IKKɛ, related to the degradation of IκB (inhibitors of NF-κB), has been studied as an antiviral innate immune regulator through its ability to control the activity of the transcription factors, IRF-3 and IRF-7 (16, 27, 34). In Mazhar's research, IKKɛ is expressed in a number of cancer cells and is involved in regulating the NF-κB activity by inducing p65/RelA phosphorylation at Ser-536 (1). In addition, IKKɛ-mediated phosphorylation of cRel leads to dissociation of the IκBα-cRel complex (15). Our findings showed that the level of IκBα increased and p-IKKɛ and p-NF-κB decreased in the HEV ORF3-infected cells, but no changes in the concentration of IKKɛ and NF-κB. Therefore, we speculated that ORF3 might inhibit the expression of p-IKKɛ through recognizing PRRs, which led to an increase in the level of IκBα following reduction of p-NF-κB, finally attenuated the NF-κB signaling pathway.
Above all, it is conceivable that HEV ORF3 diminishes the production of proinflammatory cytokines and chemotactic factors by modulating TLR4, TRAF6, and NOD2-mediated NF-κB signaling pathway, which suggests that HEV may interfere in signal transduction of macrophages by ORF3 to inhibit immune clearance of organisms, contribute to viral replication, and lead to chronic infection of HEV. Taken together, we primarily discussed the effects of HEV ORF3 on the function of macrophages and explored the mechanism, which laid the foundation for a later study.
Footnotes
Acknowledgment
This work was supported by the grant from the National Natural Science Foundation of China (No. 81300317).
Author Disclosure Statement
No competing financial interests exist.