
Abstract
CD1d-restricted T (natural killer T [NKT]) cells are important for controlling a herpes simplex virus (HSV) infection. One of the mechanisms of immune evasion by HSV is to downregulate CD1d-mediated activation of NKT cells. VP22 is an HSV-1-encoded protein responsible for reorganizing the host cell's cytoskeletal network and viral spreading. We have previously shown that modification of the cytoskeleton can alter CD1d-mediated antigen presentation. In this study, we found that an HSV-1 lacking VP22 (ΔUL49) was impaired in its ability to inhibit CD1d-mediated antigen presentation compared with the wild-type (WT) virus; this was reversed by a repair virus (UL49R) in CD1d-expressing cells. We further demonstrated that CD1d recycling was inhibited by infection with WT and UL49R, but not the ΔUL49 virus. Ectopic expression of VP22 in CD1d-expressing cells complemented the VP22-deficient virus in inhibiting antigen presentation. Moreover, inhibiting viral protein synthesis rescued VP22-dependent inhibition of CD1d antigen presentation. In conclusion, our findings suggest that VP22 is required (but not sufficient) for the inhibition of CD1d-mediated antigen presentation by an HSV-1 infection.
Introduction
H
Natural killer T (NKT) cells are a subpopulation of T cells that recognize CD1d, an MHC class I-like molecule that mediates lipid antigen presentation to NKT cells (7,18). NKT cells have been reported to play important roles in protecting a host from an HSV infection. For example, NKT cell-deficient mice are less able to clear an infection by HSV-1 or HSV-2 (3,19), although there have been reports that conflict with these observations (8). Nonetheless, in humans, two clinical cases of a life-threatening infection with the vaccine strain of varicella zoster virus (another member of the α herpesviridae subfamily), were related to a deficiency in NKT cells and/or diminished CD1d expression (5,23); these reports strongly suggest that a deficiency in the NKT cell/CD1d system is associated with increased susceptibility to herpesvirus infections. In addition, the number of NKT cells has recently been correlated with the level of HSV-1-specific antibodies in humans (34). We and others have shown that HSV-1 can inhibit CD1d-mediated antigen presentation to NKT cells by suppressing CD1d recycling (6,25,33,45). This inhibition of NKT cell function by HSV-1 is dependent on cell-to-cell contact of infected APCs and NKT cells, but not due to infection of NKT cells (6).
VP22, encoded by the UL49 gene in HSV-1, is a major tegument protein in the virion (15). A VP22-deficient virus (ΔUL49) has a defect in cell-to-cell spreading, likely due to reduced virion release (10). VP22 is also required for viral protein synthesis at late times during infection (10), and the defect in late viral protein synthesis in ΔUL49 can be rescued by secondary mutations in vhs (virion host shutoff) (28,37). During infection, VP22 is transported between cells and is necessary for the stabilization and hyperacetylation of microtubules in host cells (12,13). It forms multimers with the HSV-1-encoded proteins, V16 and VHS, and is phosphorylated by host casein kinase II and the HSV-1 viral kinase, UL13 (2,14). Furthermore, two di-leucine motifs in VP22 are important for its ability to regulate the intracellular distribution of multiple viral and cellular proteins (39).
We have previously shown that disruption of the cytoskeletal network increases CD1d-mediated antigen presentation (17). Since VP22 has an impact on the host cell's cytoskeletal network, we investigated the contribution of VP22 in the inhibition of CD1d-mediated antigen presentation following an HSV-1 infection. Our results suggest that VP22 is required (but not sufficient) for HSV-1 in the decrease of antigen presentation by CD1d postinfection.
Materials and Methods
Cell culture, constructs, and viral infections
HEK293 cells expressing wild-type (WT) CD1d and T322A/S323A mutant have been previously described (25,26). HEK293 cells were kindly provided by Prof. Philip Cohen (MRC Protein Phosphorylation Unit, University of Dundee, Scotland, United Kingdom). HeLa.CD1d cells were obtained from Dr. Janice Blum (Indiana University, Indianapolis, IN), with permission from Dr. Peter Cresswell (Yale University, New Haven, CT). Cells were cultured in DMEM supplemented with 10% FBS and 2 mM
To generate a VP22-expressing vector, two primers, 5′-AGTC
Cells were infected with the HSV-1 F strain WT virus, a VHS-deficient HSV-1 (ΔVHS), a VP22-deficient HSV-1 (ΔUL49), and rescue virus UL49R (10), for the indicated periods of time at 37°C.
Antibodies and related reagents
A pan-HSV-1- and 2-specific mAb (clone 4F10.3) was purchased from Millipore. Phycoerythrin (PE)-conjugated and purified versions of the human CD1d-specific 42.1 mAb were purchased from BD Biosciences. The pan-HLA class I-specific mAb HB95 hybridoma was kindly provided by J. Yewdell and J. Bennink (Laboratory of Viral Diseases, NIAID, NIH). The anti-CD1d free heavy chain (HC)-specific mAb clone C3D5 was from Santa Cruz Biotechnology. Recombinant cytokines (human IL-2, IL-4, and GM-CSF) were from PeproTech, Inc. Antibody pairs for the human IL-4 and GM-CSF ELISA assays were purchased from BD-Biosciences and BioLegend, respectively.
Western blotting
VP22/GFP- or GFP-expressing HEK293-CD1d cells were lysed in lysis buffer [10 mM Tris (pH 7.4), 150 mM NaCl, 0.5 mM EDTA, 2% CHAPS] and resolved on a 10% SDS-PAGE gel. The proteins were then transferred onto a PVDF membrane (Millipore). To detect VP22 expression, the membrane was incubated with a VP22-specific chicken IgY (9), followed by incubation with an HRP-conjugated anti-IgY Ab (Sigma). The membrane was then developed using chemiluminescence before exposure on film.
Flow cytometry
Cells were first washed in PBS and then fixed in 1% paraformaldehyde. To stain for surface CD1d expression, the cells were washed in Hank's balanced salt solution with 0.1% bovine serum albumin (HBSS/BSA) three times and then incubated with the 42.1 mAb. A PE-conjugated rabbit anti-mouse Ig antiserum was added as the secondary antibody. Intracellular staining with specific mAbs was performed in HBSS/BSA in the presence of 0.1% saponin. Thus, the cells were permeabilized in HBSS/BSA with 0.1% saponin and then incubated with the C3D5 mAb for detecting the CD1d HC or an HSV-specific mAb (clone 4F10.3), followed by a PE-conjugated rabbit anti-mouse Ig antiserum. All stained cells were analyzed by flow cytometry as previously described (36).
NKT cell stimulation assays
Human NKT cells were isolated from deidentified peripheral blood from normal human volunteers obtained from the Indiana Blood Center (Indianapolis, IN). The NKT cells were expanded ex vivo as described (26). Antigen-presenting cells were infected with WT or mutant HSV-1 at the indicated multiplicities of infection (MOI) and incubation time. For cycloheximide treatment, cells were treated with medium control or cycloheximide (25 μg/mL) for 30 min. The cells were then infected with the viruses as indicated in the presence or absence of cycloheximide and subsequently fixed in 0.05% paraformaldehyde, washed three times with RPMI medium (Lonza) supplemented with 10% FBS, and cocultured with human NKT cells as previously described (25). Cytokine (IL-4 and GM-CSF) production by NKT cells was measured by ELISA.
Recycling assay
The recycling assay was performed as previously described (25,45). Briefly, 5 million HEK293-CD1d WT cells were infected with WT or mutant HSV-1 (MOI = 2) for 4 h at 37°C. One hour before harvest, cyclohexamide (25 μg/mL) was added. Purified 42.1 mAb was added to block surface CD1d. After washing, the cells were kept in IMDM medium (Lonza) containing cyclohexamide for different time intervals at 37°C. At the indicated time points, an aliquot was placed in a well of an ice-cold 96-well plate. All cells were stained with a PE-labeled CD1d-specific mAb (42.1) on ice. After washing three times in ice-cold PBS, the cells were fixed in 1% paraformaldehyde and resuspended in HBSS/BSA buffer until data acquisition using flow cytometry.
Statistical analysis
Graphs were made and statistics analyzed using GraphPad Prism 5 (GraphPad Software). Each experiment was performed at least three times and one representative assay is shown. The error bars represent the standard error of the mean (SEM) of triplicate samples. A two-way ANOVA, one-way ANOVA with Bonferroni's post-test, or Student's t-test was used as appropriate. A p-value of less than 0.05 was considered significant.
Results
VP22 is required for the inhibition of antigen presentation following an HSV-1 infection
Previous work from our laboratory has demonstrated that the disruption of actin filaments increases CD1d-mediated antigen presentation (17). Conversely, TGF-β treatment of CD1d-expressing cells, which induces the formation of actin stress fibers, inhibits antigen presentation by murine (4) and human CD1d (Supplementary Fig. S1; Supplementary Data are available online at
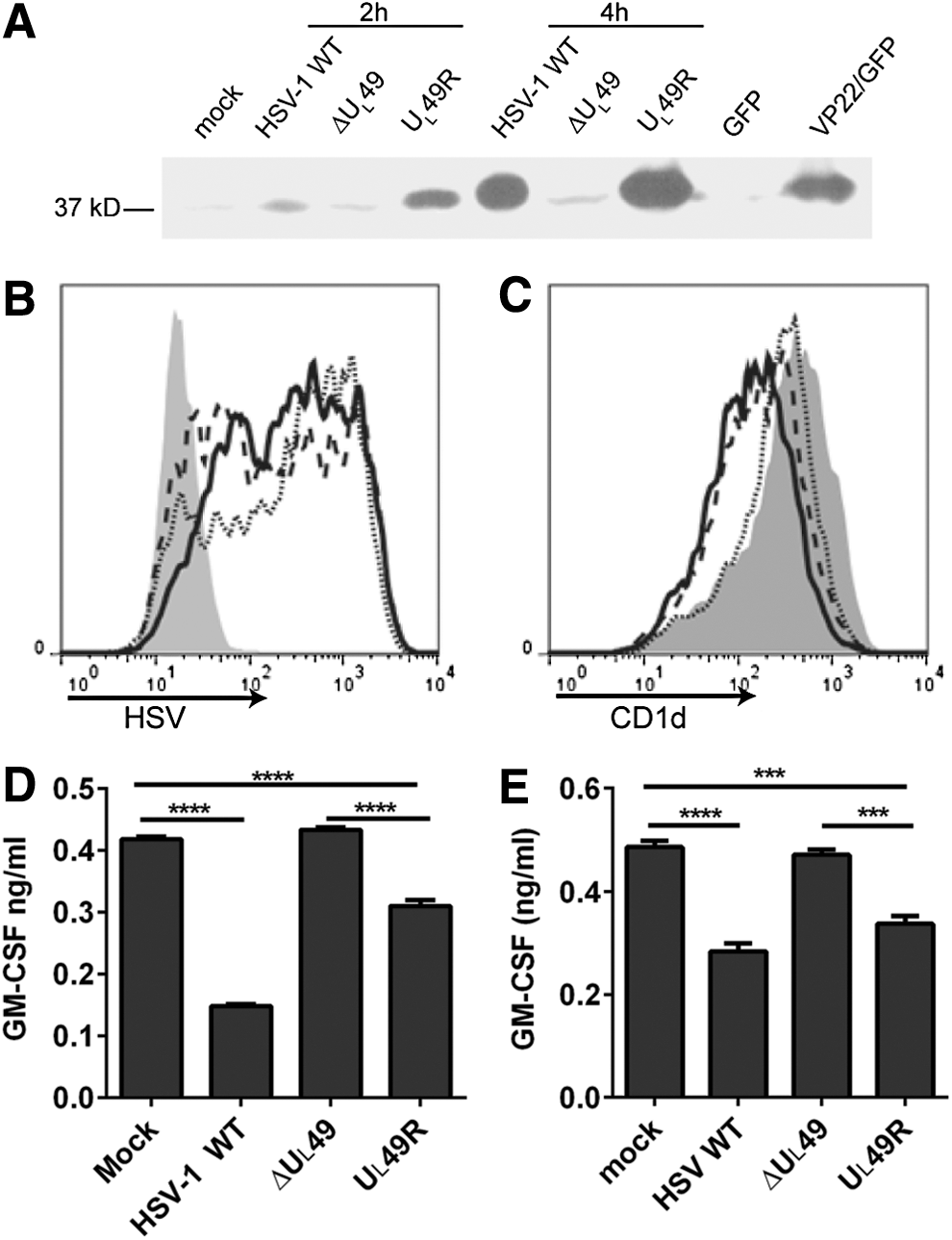
VP22 is required for the inhibition of CD1d-mediated antigen presentation to NKT cells.
VP22 is important for inhibiting CD1d recycling upon an HSV-1 infection
It was previously reported that an HSV-1 infection inhibits CD1d-mediated antigen presentation, likely by inhibiting CD1d molecules from recycling back to the cell surface (25,45). VP22 is known to modify the cytoskeletal network in host cells (13), which may cause changes in the endocytic trafficking of cellular proteins. To determine whether CD1d trafficking is affected by VP22, recycling assays were performed on HEK293-CD1d cells that were infected for 4 h with WT HSV-1, ΔUL49, or UL49R. The infected cells expressed similar levels of HSV viral proteins (Fig. 2A). Consistent with previous reports (6,25,33,45), an HSV-1 infection reduced the ability of CD1d molecules to recycle back to the cell surface (Fig. 2B). In cells infected with ΔUL49, CD1d recycled at a similar rate as those in mock-treated cells, and therefore, much faster than CD1d in cells infected with either UL49R or the WT HSV-1 (Fig. 2B). Thus, these data confirm the observation that CD1d molecules are impaired in recycling back to the surface following an HSV-1 infection and moreover, show that this is VP22 dependent.
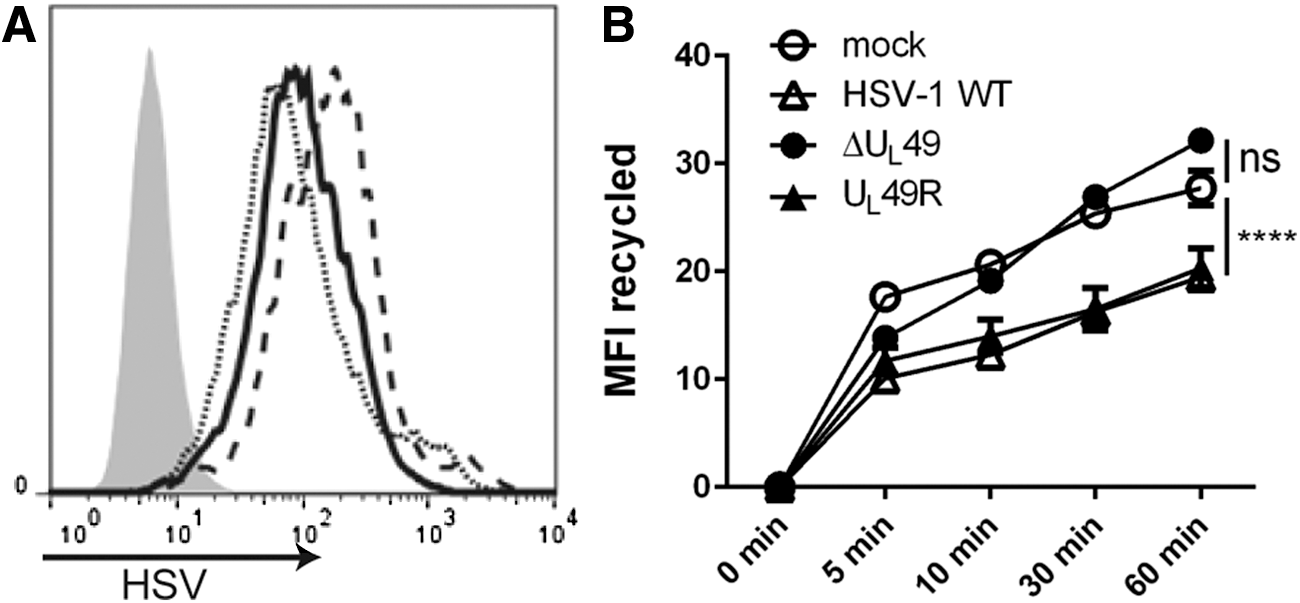
VP22 is important for inhibiting CD1d recycling upon an HSV-1 infection. HEK293-CD1d cells were mock treated (gray-filled histogram) or infected with WT HSV-1 (solid line), ΔUL49 (dotted line) or UL49R (dashed line) viruses at an MOI of 2 for 4 h
VP22 is not sufficient for inhibiting CD1d-mediated antigen presentation postinfection
To determine if VP22 is sufficient for the inhibition of CD1d-mediated antigen presentation that was observed post-HSV infection, VP22 cDNA was stably transfected into HEK293-CD1d cells using a GFP-tagged bicistronic IRES construct (HEK293-CD1d.VP22/GFP cells). The ectopic expression of VP22 in HEK293-CD1d.VP22/GFP cells was confirmed by western blot analysis (Fig. 3A). We found that CD1d expression in both control and VP22-expressing HEK293-CD1d cells was comparable (Fig. 3B). The cells were also similar in terms of antigen presentation to NKT cells (Fig. 3C). These results suggest that the ectopic expression of VP22 is not sufficient to inhibit CD1d-mediated antigen presentation to NKT cells, as we observed following infection with WT HSV-1.
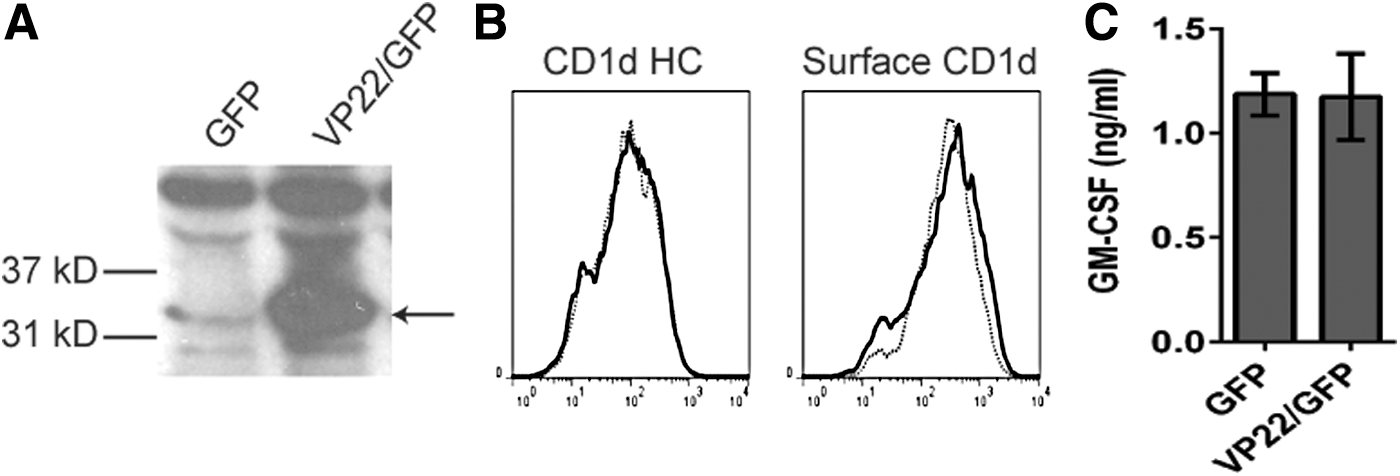
VP22 is not sufficient to inhibit antigen presentation by CD1d following an HSV-1 infection.
Ectopic expression of VP22 complements the ΔUL49 virus in inhibiting CD1d-mediated antigen presentation
Since ectopic expression of VP22 did not, by itself, inhibit CD1d-mediated antigen presentation (Fig. 3C), we hypothesized that other viral proteins (in the context of the whole virion) contribute to the inhibition of antigen presentation observed postinfection. To test this hypothesis, HEK293-CD1d.VP22/GFP cells and control cells were infected with the VP22-deficient (ΔUL49) or VP22-rescued (UL49R) HSV-1. For cells infected with the ΔUL49 virus, ectopic expression of VP22 resulted in a significantly greater decrease in antigen presentation by CD1d, as compared with cells expressing a control vector (Fig. 4C). For cells infected with the UL49R virus, VP22 expression in host cells also caused a further inhibition in antigen presentation (Fig. 4C). Similar viral infectivity in these cells was confirmed by flow cytometry using an HSV-specific antibody (Fig. 4A). Furthermore, we did not observe any noticeable changes in CD1d surface expression postinfection (Fig. 4B). Therefore, these data suggest that VP22 requires coexpression of other HSV-1-encoded proteins in the inhibition of antigen presentation by CD1d postinfection.
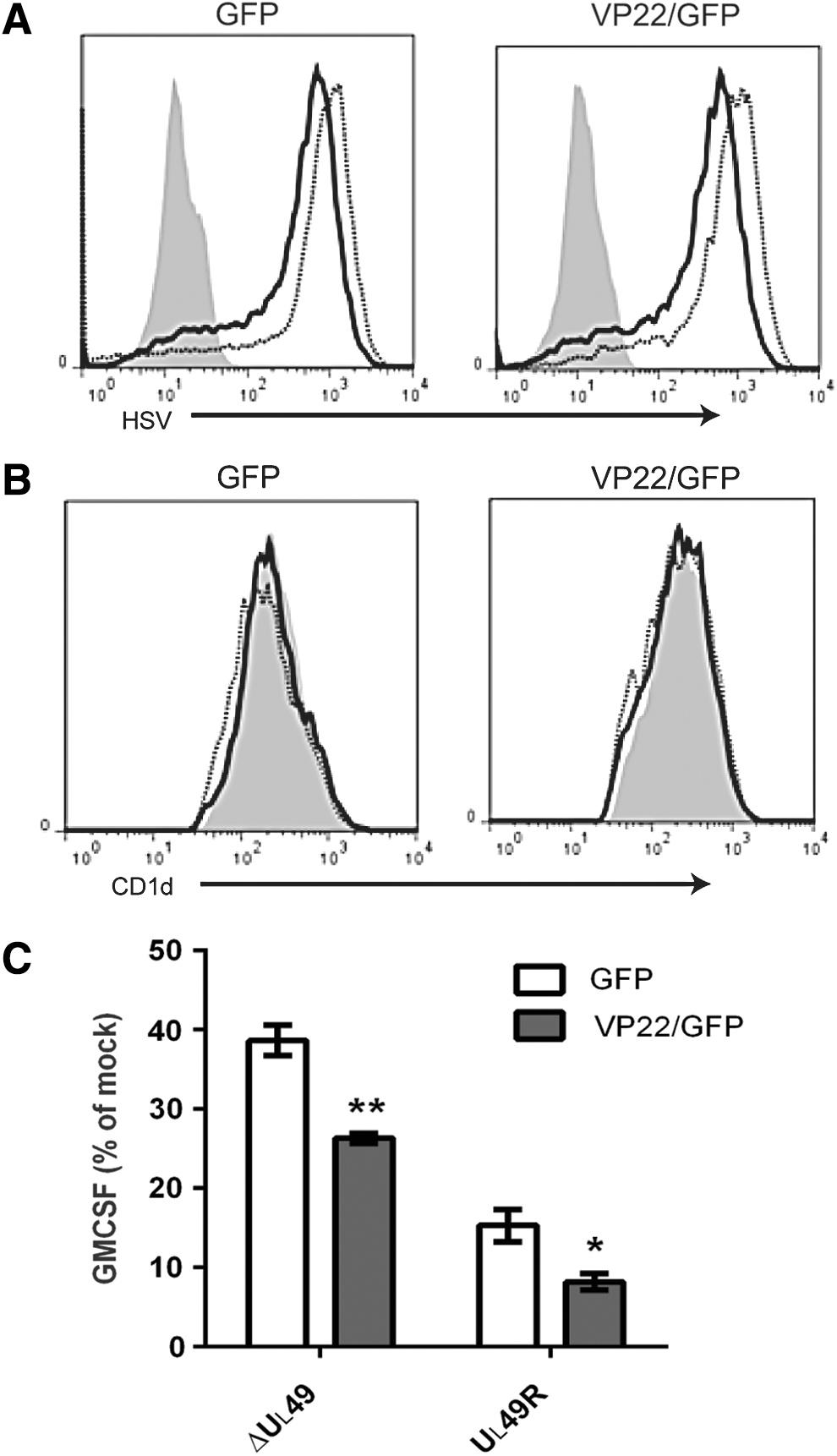
Ectopic expression of VP22 complements ΔUL49 virus in the inhibition of antigen presentation following an HSV-1 infection.
Viral protein synthesis is important for VP22-mediated inhibition of antigen presentation
To determine whether other viral proteins are important for the observed VP22-dependent inhibition of CD1d Ag presentation, we utilized the protein synthesis inhibitor, cycloheximide. We found that cycloheximide treatment by itself had a minimal effect on CD1d-mediated antigen presentation in HEK293-CD1d cells (data not shown). Interestingly, cycloheximide treatment in WT HSV-1-infected HEK293-CD1d.GFP cells rescued CD1d-mediated antigen presentation (Fig. 5), suggesting that viral protein synthesis inhibits CD1d Ag presentation. On the other hand, HEK293-CD1d.VP22/GFP cells had similar CD1d Ag presentation as control cells when cells were infected with HSV-1 WT virus, with or without cycloheximide treatment. When HEK293-CD1d.VP22/GFP cells were infected with the ΔUL49 HSV-1 virus, they were less able to activate NKT cells compared with control cells (Fig. 5). In contrast, when HEK293-CD1d.VP22/GFP cells were treated with cycloheximide and infected with ΔUL49, there was enhanced CD1d-mediated antigen presentation compared with control cells. Therefore, our data suggest that the synthesis of other viral proteins is also important for the VP22-mediated inhibition of antigen presentation by CD1d.
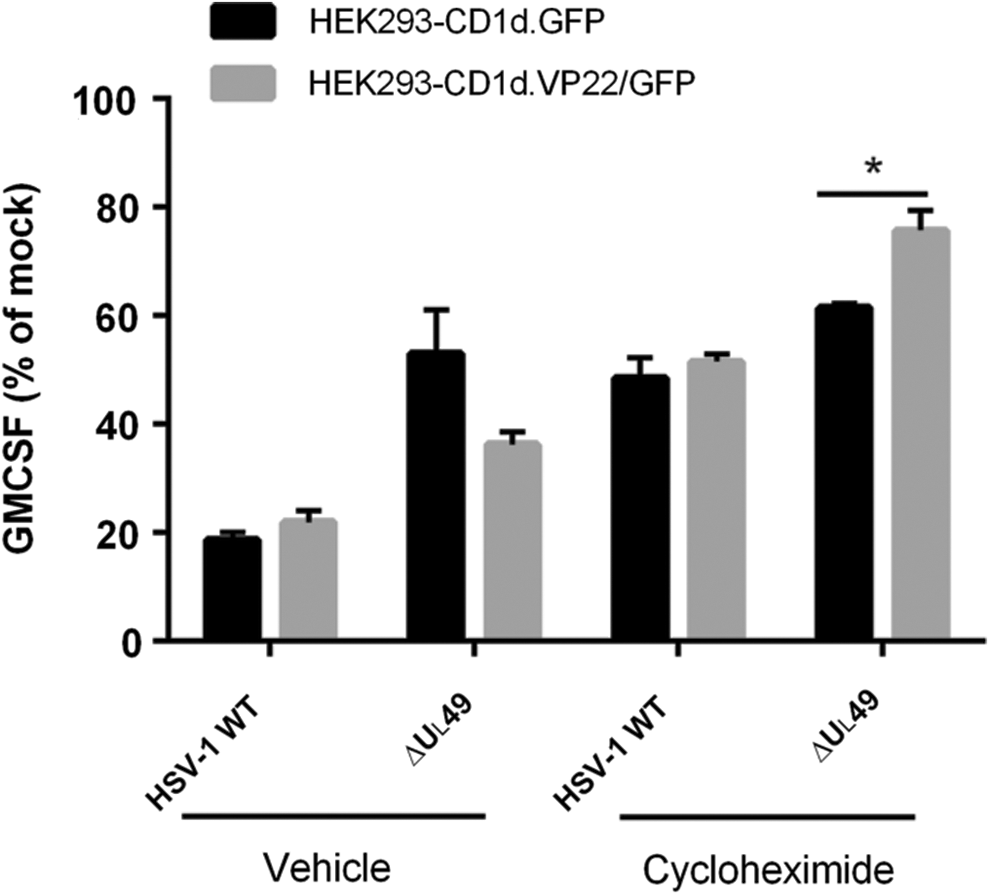
Viral protein synthesis is important for VP22-mediated inhibition of CD1d-mediated antigen presentation. HEK293-CD1d (VP22/GFP) and control (GFP) cells were mock infected or infected with ΔUL49 for 6 h at an MOI of 5, with or without cycloheximide. The cells were subsequently fixed and cocultured with human NKT cells for 48 h. Data are presented as the percentage of GM-CSF produced by NKT cells relative to mock treatment (set at 100%). Representative data from three independent experiments are shown. *p < 0.05 (Student's t-test).
Discussion
VP22 is a major tegument protein in the HSV-1 virion and has many important functions that are critical for HSV-1 replication and pathogenicity (39). In the current study, we have shown that VP22 is necessary (but not sufficient) for inhibiting CD1d-mediated antigen presentation following an HSV-1 infection. It is likely that VP22 requires the presence of other HSV-1-encoded proteins to be functional. For example, in HSV-1-infected cells, VP22 forms homotypic complexes, which then associate with the HSV-1-encoded proteins, VP16 and UL13 (29). VP22 also forms a complex with VP16, which interacts with VHS; the presence of both VP22 and VP16 is required for the accumulation of VHS (38). It is known that in the absence of VP22, the expression of other viral proteins is also impaired. For example, less expression of ICP0, VP16, VHS, glycoprotein D, and gE has been reported in VP22-null mutant viruses (11). VP22 is also required for optimal expression of the HSV-1-encoded protein, gB (39). Interestingly, gB has been shown to bind CD1d and alter CD1d-mediated antigen presentation postinfection (35). Nonetheless, gB itself is not sufficient to inhibit CD1d-mediated antigen presentation (35). A recent publication indicates that KIF3A, a subunit of kinesin-2 motor protein that transports protein complexes along microtubule tracks, is required for CD1d surface expression (43). KIF3A can be phosphorylated by the viral kinase US3 and downregulates CD1d surface expression (43). This is consistent with our discovery that VP22, a viral protein modifies host cell microtubule network, is important in inhibiting CD1d-mediated antigen presentation.
VP22 is highly phosphorylated and it can be phosphorylated by both viral and host kinases (14). Although the phosphorylation profiles of VP22 synthesized during transfection and infection are the same, ectopically expressed VP22 lacks the nonphosphorylated form of VP22, which exists in infected cells. Nonphosphorylated VP22 is the major form of VP22 in virions (14), and in infected cells it exhibits enhanced binding to microtubule bundles and efficiently interacts with VP16 (32). It is possible that the nonphosphorylated form of VP22, absent in VP22-transfected cells, is the functional form that inhibits CD1d recycling and antigen presentation. This may explain why ectopic expression of VP22 in HEK293-CD1d cells does not inhibit CD1d-mediated antigen presentation. Inhibiting viral protein synthesis using cycloheximide abolishes the inhibition of CD1d-mediated antigen presentation by ectopically expressed VP22 (Fig. 5), suggesting other viral proteins cooperate with (or assist) VP22, allowing for its effect on CD1d. Further investigations are needed to determine whether the different forms of VP22 are functionally distinct in host cells.
A VP22-deficient virus (ΔUL49) has been reported to produce a smaller plaque and there is reduced virion release in infected cells (10). One may argue that the reduced inhibition in antigen presentation observed following infection with ΔUL49 is due to its impaired infectivity. However, the ΔUL49 virus did not show a significant difference from WT HSV-1 or a VP22-rescued virus (UL49R) in single cycle growth analyses when cells were infected at a high multiplicity of infection (10). Indeed, our data also demonstrated that the ΔUL49 virus was just as infectious as WT and UL49R viruses 4 h postinfection (Fig. 1A). In addition, infection of HEK293-CD1d cells with another HSV-1 mutant virus (ΔUS3/ΔUL13), which also produces very small plaques, inhibited CD1d-mediated antigen presentation at a similar level as WT HSV-1 (25). Consistently, infection with the ΔUS3/ΔUL13 virus also inhibited CD1d recycling in HEK293-CD1d cells (data not shown). Therefore, the reduced ability of the ΔUL49 virus to inhibit CD1d-mediated antigen presentation is not due to reduced infectivity.
We and others have reported that an HSV-1 infection prevents intracellular CD1d from recycling back to the cell surface (25,45). Our study has found that VP22 is required for the inhibition of CD1d recycling postinfection. To the best of our knowledge, it had not been previously shown that VP22 is directly involved in regulating subcellular vesicular trafficking. However, VP22 has been reported to bind to microtubule bundles and myosin HC 9, the latter being a component of the actin cytoskeleton (13,32). Similarly, substitution of the CD1d residues T322 and S323 with Ala (T322A/S323A) prevents their phosphorylation and also increases the CD1d recycling rate postinfection (25). In a previous study, we speculated that viral infections may activate host Ser/Thr kinases and thereby phosphorylate the cytoplasmic tail of CD1d, causing lysosomal targeting and degradation of CD1d (25). Further studies will be important to determine how VP22 impacts these activities post-HSV-1 infection.
In summary, our current study has demonstrated that following an HSV-1 infection, VP22 is required (but not sufficient) for the inhibition of CD1d recycling and antigen presentation to NKT cells. Future studies will be focused on how VP22 may interact with other viral proteins and the host cell's cytoskeletal network and, as a result, regulate the functional expression of CD1d.
Footnotes
Acknowledgments
The authors would like to thank Drs. P. Cohen (University of Dundee), P. Cresswell (Yale University), and J. Blum (Indiana University) for critical reagents, and technical assistance from the Flow Cytometry Resource Facility, Indiana University School of Medicine. This work was supported by the National Institutes of Health grants R01 AI46455 and P01 AI056097 (to R.R.B.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.