
Abstract
Association between variable agent-induced hepatocellular carcinoma (HCC) and both PAI-1 4G/5G polymorphism and plasminogen activator inhibitor (PAI-1) levels compared to healthy controls have been reported in earlier studies. We aimed to assess serum PAI-1 and PAI-1 4G/5G polymorphism in hepatitis C virus (HCV)-induced HCC, HCV-induced liver cirrhosis, and viral infection-free apparently healthy control subjects. Forty nine HCC, 52 cirrhosis, and 105 controls were genotyped for PAI-1 4G/5G using an allele-specific polymerase chain reaction analysis. In addition, for 31 HCC, 24 cirrhosis, and 28 controls, serum PAI-1 level was measured by enzyme-linked immunosorbent assay (ELISA). There was no significant difference in PAI-1 4G/5G genotype distribution between cirrhosis and controls (p = 0.33, p = 0.15, and p = 0.38 for the codominant, dominant, and recessive models, respectively) or between HCC and cirrhosis (p = 0.5, p = 0.24, and p = 0.69 for the codominant, dominant, and recessive models, respectively). Serum PAI-1 was significantly higher in cirrhosis than controls and significantly lower in HCC than cirrhosis (p < 0.001 for both). Serum PAI-1 did not differ significantly among the three PAI-1 4G/5G genotypes in controls, cirrhosis, and HCC (p = 0.29, p = 0.28, and p = 0.73 respectively). We documented higher serum PAI-1 in HCV-induced HCC than viral infection-free controls, but interestingly, lower than HCV-induced liver cirrhosis patients. This was not genotype related. Further studies will be needed to clearly elucidate the underlying mechanism.
Introduction
P
Hepatocellular carcinoma (HCC) is one of the long-term complications of hepatitis C virus (HCV) infection. In most cases, HCV-induced HCC is the last step of a progressive course from hepatitis to HCC (12). Although aspects of this pathogenesis have been investigated, its underlying mechanisms are still evasive. HCV harbors an RNA genome that does not integrate in the host cell genome (1). As a result, indirect mechanisms of HCV-induced hepatocarcinogenesis have been suggested.
Studies (4,5) have reported an elevated level of PAI-1 in variable agent-induced HCC compared to healthy controls, which was more obvious with viral-induced HCC. Weng et al. (14) and Divella et al. (5) reported an association between variable agent-induced HCC risk and PAI-1 4G/5G polymorphism (compared to healthy controls) with special association with viral-induced HCC in Divella et al. study (5). Divella et al. (3) indicated PAI-1 genetic variations as a prognostic marker for HCC.
The link of serum PAI-1 and PAI-1 4G/5G polymorphism to HCV-induced HCC in relation to HCV-induced cirrhosis as a leading step in hepatocarcinogenesis is poorly investigated. In this study, we aimed to assess serum PAI-1 and PAI-1 4G/5G polymorphism in HCV-induced HCC, HCV-induced liver cirrhosis, and viral infection-free apparently healthy control subjects. To the best of our knowledge, PAI-1 4G/5G polymorphism in HCV-induced HCC compared with that in HCV-induced cirrhosis was not addressed before.
Subjects and Methods
Study population
The study comprised 52 patients with HCV-induced liver cirrhosis, 49 patients with HCV-induced HCC, and 105 viral infection-free apparently healthy control subjects. They were all Egyptians, an ethnic group in North Africa. Patients with cirrhosis were in the age of 53.02 ± 6.84 years. They were 34 males and 18 females. Patients with HCC were in the age of 55.43 ± 8.91 years. They were 39 males and 10 females. Control subjects were in the age of 54.07 ± 6.17 years. They were 80 males and 25 females. Patients and controls were age matched (p = 0.33 for cirrhosis/controls, p = 0.13 for HCC/cirrhosis, and p = 0.27 for HCC/controls). Patients and controls were gender matched (p = 0.15 for cirrhosis/controls, p = 0.11 for HCC/cirrhosis, and p = 0.64 for HCC/controls). All patients were diagnosed on the basis of history, clinical, laboratory (including HCV antibodies, HCV-RNA polymerase chain reaction (PCR), and alpha feto protein (AFP), and radiological examinations and confirmed by liver biopsy. Informed consent was obtained from all individual participants included in the study before blood and data collection. The protocol and procedures followed were in accordance with the ethical standards of the responsible institutional committee on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
PAI-1 4G/5G genotyping assays
High molecular weight DNA was extracted from peripheral blood using DNA extraction kit (QIAamp®DNA Blood Mini Kit; Qiagen). DNA was quantified spectrophotometrically and stored. Isolated DNA was amplified for PAI-1 4G/5G insertion/deletion polymorphism, located within the promoter region −675 bp upstream of the transcription start site, using an allele-specific PCR. Primers used (10) are as follows: control upstream (PAI-1): 5′ AAG CTT TTA CCA TGG TAA CCC CTG GT 3′, common downstream (PAI-2): 5′ TGC AGC CAG CCA CGT GAT TGT CTA 3′, deletion 4G allele (PAI-4G): 5′ GTC TGG ACA CGT GGG GA 3′, and insertion 5G allele (PAI-5G): 5′ GTC TGG ACA CGT GGG GG 3′. PAI-1 and PAI-2 amplify a 257-bp product from either allele. The 4G allele was specifically amplified using PAI-4G, and the 5G allele was specifically amplified using PAI-5G. PCRs were performed in 25 μL volumes containing 1 μg/mL DNA, both PAI-1 and PAI-2, and either PAI-4G or PAI-5G in Go Taq® Green Master Mix, 2X (Promega corporation). Thermal cycling conditions starts with a 94°C hold for 5 min followed by 40 cycles, each consists of 1 min denaturation at 94°C, 45 s annealing at 58°C, and 1 min extension at 72°C. An extension for 15 min at 72°C was added, followed by a 4°C hold. The amplified products were visualized using ethidium bromide-stained agarose gel electrophoresis. For every PCR run, a nontemplate control was included. Representative gel is shown in Figure 1.
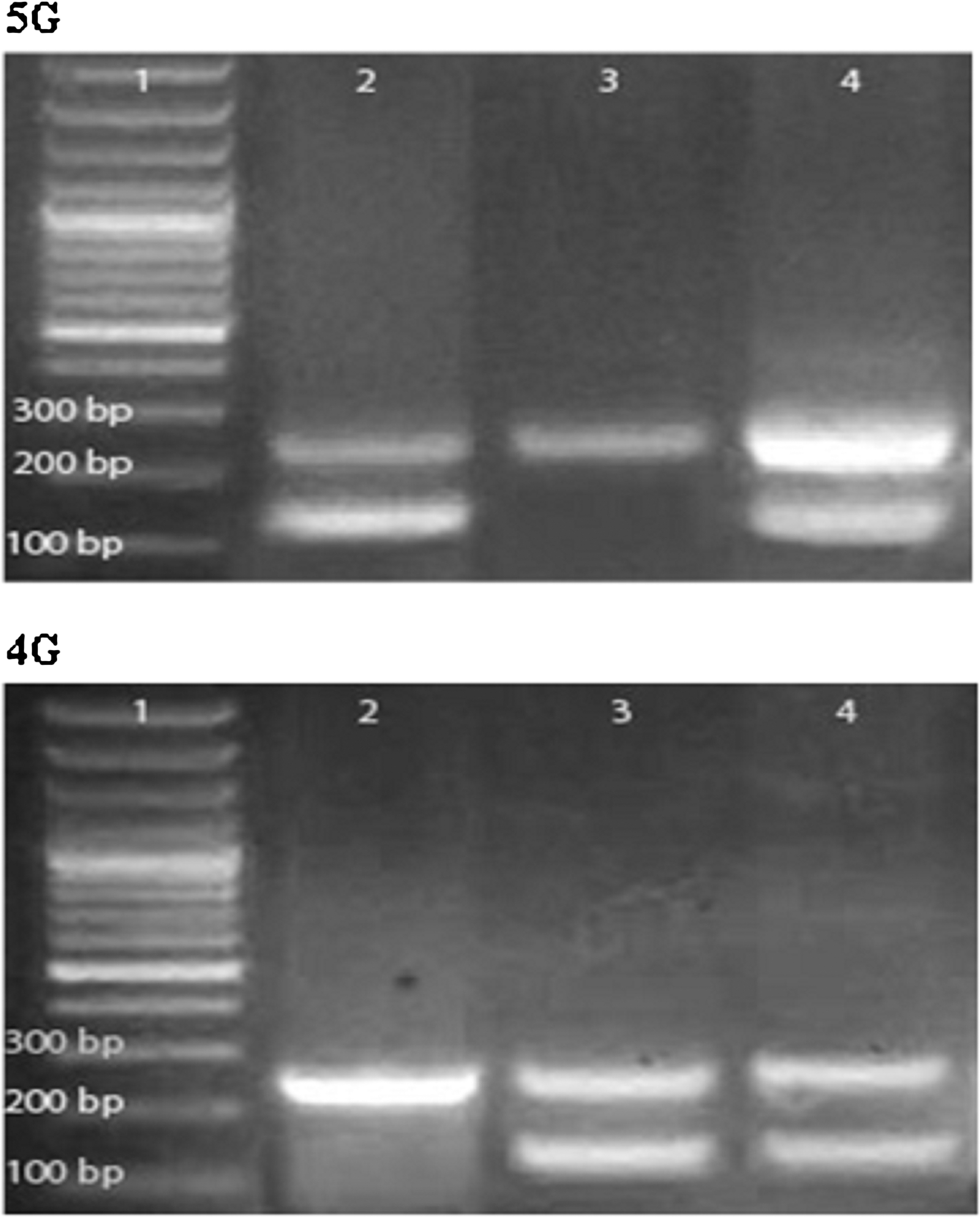
2% agarose gel electrophoresis for 5G (top) and 4G (bottom) polymerase chain reaction products. Lanes 1 (top and bottom) are 100-bp molecular weight markers. Lanes 2 (top and bottom) represent 5G/5G genotype. Lanes 3 (top and bottom), represent 4G/4G genotype. Lanes 4 (top and bottom) represent 4G/5G genotype.
Serum PAI-1 measurement
Blood samples were collected in red capped plain vacutainer tubes. Sera were separated and part of each was freezed (−20) until Human PAI-1 measurement. Sera were available for 31 HCC patients, 24 liver cirrhosis patients, and 28 controls.
Human PAI-1 was measured using the Human PAI-1 invitrogen enzyme-linked immunosorbent assay (ELISA) kit according to manufacturer's instructions. This is a solid-phase sandwich ELISA designed to detect and quantify the level of human PAI-1 in serum, EDTA and heparin plasma, buffered solution, and tissue culture medium. The assay recognizes both natural and recombinant human PAI-1. According to the kit data-sheets,
(i) Intra-assay Precision: Samples of known human PAI-1 content were assayed in replicates of 14 to determine precision within an assay. Sample 1 (mean: 339.82 pg/mL, standard deviation: 12.49, % coefficient of variation: 3.68), sample 2 (mean: 982.59 pg/mL, standard deviation: 48.66, % coefficient of variation: 4.95), and sample 3 (mean: 2414.94 pg/mL, standard deviation: 103.23, % coefficient of variation: 4.27).
(ii) Interassay precision: Samples were assayed 42 times in multiple assays to determine precision between assays. Sample 1 (mean: 361.65 pg/mL, standard deviation: 32.73, % coefficient of variation: 9.05), sample 2 (mean: 961.19 pg/mL, standard deviation: 76.83, % coefficient of variation: 7.99), and sample 3 (mean: 2466.57 pg/mL, standard deviation: 150.23, % coefficient of variation: 6.09).
(iii) Analytical sensitivity: The minimum detectable dose of human PAI-1 is <30 pg/mL.
(iv) Linearity of dilution: Linear regression analysis of samples versus the expected concentration yielded average correlation coefficients of 0.999 for serum.
(v) Average % recovery for human serum: 94.
(vi) Standards: 2,000; 1,000; 500; 250; 125; 62.5; and 0 pg/mL Hu PAI-1.
(vii) Expected values for human serum: 320–8,560 pg/mL.
Statistical analysis
Qualitative data are presented as numbers and percentages. Quantitative data are presented as means ± standard deviation. Chi-squared test was used to compare the proportions of categorical variables. Student t-test was used to compare quantitative parametric variables. Kruskal–Wallis and Mann–Whitney tests were used to compare quantitative nonparametric variables. The distributions of genotypes for PAI-1 4G/5G gene were tested for the Hardy–Weinberg heredity equilibrium by exact test. Odds ratios and 95% confidence intervals were calculated to examine the association between the PAI-1 4G/5G genotypes and the risk. The codominant, the dominant, and the recessive models were used. p < 0.05 was considered significant.
Results
A total of 206 subjects (52 patients with HCV-induced liver cirrhosis, 49 patients with HCV-induced HCC, and 105 viral infection-free apparently healthy control subjects) were enrolled in the study. Demographic, clinical, and laboratory characteristics of our patients are listed in Table 1.
HCC, hepatocellular carcinoma; HCV, hepatitis C virus; SD, standard deviation.
Genotype distribution in viral infection-free controls, HCV-induced liver cirrhosis, and HCV-induced HCC did not deviate from the Hardy–Weinberg equilibrium (p = 0.69, p = 0.54, and p = 1 respectively) (Table 2).
PAI-1, plasminogen activator inhibitor.
The frequency distribution of PAI-1 4G/5G polymorphism is presented in Table 3. In the codominant, dominant, and recessive models, no significant difference in genotype distribution was observed between viral infection-free controls and HCV-induced liver cirrhosis (p = 0.33, p = 0.15, and p = 0.38, respectively). In the codominant, dominant, and recessive models, no significant difference in genotype distribution was observed between HCV-induced liver cirrhosis and HCV-induced HCC patients (p = 0.5, p = 0.24, and p = 0.69, respectively).
CI, confidence interval; OR, Odds ratio.
The frequency distribution of PAI-1 5G and 4G alleles did not differ significantly between controls and cirrhosis patients (p = 0.14) or cirrhosis and HCC patients (p = 0.29) (Table 4).
Serum PAI-1 was significantly higher in HCV-induced liver cirrhosis than viral infection-free controls and significantly lower in HCV-induced HCC than HCV-induced liver cirrhosis (p < 0.001 for both). It was significantly higher in HCC than controls (p = 0.002). In carriers of 5G/5G genotype, serum PAI-1 was significantly higher in cirrhosis than controls and significantly lower in HCC than cirrhosis (p < 0.001 for both). In carriers of 5G/4G genotype, Serum PAI-1 was significantly higher in cirrhosis than controls and significantly lower in HCC than cirrhosis (p = 0.005 and p = 0.01, respectively). In carriers of 4G/4G genotype, serum PAI-1 was significantly higher in cirrhosis than controls and significantly lower in HCC than cirrhosis (p = 0.001 and p = 0.004, respectively) (Table 5).
Bold P values are statistically significant.
Controls/cirrhosis.
Cirrhosis/HCC.
Controls/HCC.
Serum PAI-1 did not differ significantly among the three PAI-1 4G/5G genotypes in controls, cirrhosis, and HCC (p = 0.29, p = 0.28, and p = 0.73, respectively) (Table 6).
Discussion
In this study, we investigated PAI-1 4G/5G polymorphism and serum PAI-1 in HCV-induced HCC and HCV-induced cirrhosis; as a leading step in hepatocarcinogenesis and in viral infection-free control subjects. Our analysis revealed that there was no significant difference in PAI-1 4G/5G genotype distribution between viral infection-free controls and HCV-induced liver cirrhosis. Similarly, D'Amico et al. (2) reported a substantial role of PAI-1 4G/4G in patients with alcohol and cryptogenic liver cirrhosis, in a Caucasian population, but not in patients with HCV or hepatitis B virus (HBV). Weng et al. (14) and Divella et al. (5) reported an association between variable agent-induced HCC risk and PAI-1 4G/5G polymorphism with special association with viral-induced HCC in Divella et al. (5) study. These studies make use of healthy controls. However, as HCV-induced HCC develops after decades of infection, this strategy might not prove to be adequate. Therefore, in this study, we used HCV-infected noncancerous patients (patients with cirrhosis; a leading step in hepatocarcinogenesis) with the aim to identify carcinogenic risk estimation in HCV-infected patients. Our results demonstrated that there was no significant difference in PAI-1 4G/5G genotype distribution between HCV-induced liver cirrhosis and HCV-induced HCC patients. However, ethnicity differences in the observation groups could be considered, as polymorphisms vary depending on the geographical distribution and ethnicity.
Our results revealed that serum PAI-1 was significantly higher in HCV-induced liver cirrhosis than viral infection-free controls. Similar results were obtained for plasma PAI-1 in two earlier studies (4,7).
Divella et al. (4) studied plasma PAI-1 in patients with HCC induced by the presence of both viruses (HCV and HBV), patients with HCC induced only by the HCV virus, and patients with HCC free from viral infection. Significantly, higher levels of PAI-1 were reported when virus infection, HCV only or both HCV and HBV, was present. One striking observation that emerged from our results was that serum PAI-1 was significantly lower in HCV-induced HCC patients than HCV-induced liver cirrhosis, despite being significantly higher in HCC than viral infection-free controls. Interestingly, this was observed on analyzing each genotype separately. However, higher serum PAI-1 in HCC than controls reached statistical significance in 4G/4G genotype only (not in 5G/5G or 4G/5G genotypes). Divella et al. (5) found that patients with the 4G/4G genotype had a significantly higher plasma level of PAI-1 when compared to patients with the 4G/5G genotype and patients with the 5G/5G genotype. In contrast, our results demonstrated that serum PAI-1 level did not differ significantly among PAI-1 4G/5G genotypes within controls, cirrhosis, or HCC patients. In HCC, the mean serum PAI-1 level was higher in the 4G/4G genotype, but it did not reach statistical significance. These conflicting observations could be attributed to heterogeneity of the variable agent-induced HCC patients included in Divella et al. study (5). The difference in biospecimens (serum/plasma) should be considered. Moreover, in their study, (5) HBV can affect activation of the promoter of serpin-1; probably being a DNA virus, it integrates into the host genome and acts directly on the promoter of the gene, in turn, influencing the circulating levels of PAI-1. However this is not the case for HCV.
Lack of association between PAI-1 4G/5G polymorphism and risk of cirrhosis or risk of HCC in HCV-infected patients, together with higher serum PAI-1 in both cirrhosis and HCC patients, indicate that HCV-induced cirrhosis may be the reason for higher levels of serum PAI-1 and not necessarily the HCC, and serum PAI-1 can have a role in hepatocarcinogenesis in HCV-infected patients that is mostly not related to PAI-1 4G/5G genotypes. Matsuzaki et al. (11) results indicated that interleukin-1β activates c-Jun N-terminal kinase (JNK) that stimulates the pSmad3L/PAI-1 pathway in facilitating hepatocytic invasion, meanwhile decreasing the TGF-β-dependent tumor-suppressive activity by the pSmad3C/p21(WAF1) pathway. As a result, chronic inflammation associated with HCV infection directs hepatocytic TGF-β signaling from tumor suppression to fibrogenesis, enhancing liver fibrosis and HCC risk. HCV infection itself or the intracellular expression of HCV genes might act by modifying the host cell methylation pattern, however, further studies will be needed to clarify the underlying mechanisms.
Conclusion
Our results documented higher serum PAI-1 in HCV-induced HCC than viral infection-free controls, but interestingly, lower than HCV-induced liver cirrhosis patients. Lack of association between PAI-1 4G/5G polymorphism and risk of cirrhosis or risk of HCC in HCV-infected patients together with higher serum PAI-1 in both cirrhosis and HCC patients than viral infection-free controls raises the need for studies that explore the underlying mechanisms of PAI-1 overexpression, its role in hepatocarcinogenesis, and hence in treatment direction in HCV-infected patients.
Footnotes
Author Disclosure Statement
No competing financial interests exist.