
Abstract
Impairment of immune defenses can contribute to severe influenza infections. Rapamycin is an immunosuppressive drug often used to prevent transplant rejection and is currently undergoing clinical trials for treating cancers and autoimmune diseases. We investigated whether rapamycin has deleterious effects during lethal influenza viral infections. We treated mice with two concentrations of rapamycin and infected them with A/Puerto Rico/8/1934 (A/PR8), followed by a heterosubtypic A/Hong Kong/1/68 (A/HK68) challenge. Our data show similar morbidity, mortality, and lung viral titer with both rapamycin treatment doses compared to untreated controls, with a delay in morbidity onset in rapamycin high dose recipients during primary infection. Rapamycin treatment at high dose also led to increase in percent cytokine producing T cells in the spleen. However, all infected animals had similar serum antibody responses against A/PR8. Post-A/HK68 challenge, rapamycin had no impeding effect on morbidity or mortality and had similar serum antibody levels against A/PR8 and A/HK68. We conclude that rapamycin treatment does not adversely affect morbidity, mortality, or antibody production during lethal influenza infections.
Introduction
A
Among different types of immunosuppression, the drug-induced form of immunosuppression has the potential to make patients more susceptible to severe influenza illness (6,20) and is, therefore, an important public health issue that needs to be investigated. Some studies using animal models have, however, not shown such compromising effects during infections in conjunction with immunomodulatory therapies. A study using rosuvastatin showed no difference in influenza A infection clinical course and viral replication (26). Others have shown ameliorative effects through suppression of cytokine storm (30) and inhibition of Nox2 oxidase activity (29). Inhibition of the mammalian target of rapamycin (mTOR) has also improved immunological outcomes in experimental animals (1,11,17,24) and clinically (21). In addition, inhibitors of mTOR have been shown to be beneficial in the absence of infection, such as with influenza vaccine response in humans (31) and increasing lifespan in mice (4,16).
Rapamycin (Sirolimus) was approved in 1999 by the FDA to prevent transplant rejection by inhibiting mTOR (10) and is currently in clinical trials for treating a variety of different cancers and autoimmune diseases, highlighting the broad scope of its therapeutic effects (14,23,33). mTOR is a key member of a cellular pathway that senses the environment for optimal cell proliferation. The specific target for rapamycin, the mTORC1 complex, is responsible for two main phosphorylation events with p70 ribosomal S6 kinase (p70S6K) and the binding protein eukaryotic translation initiation factor eIF4E (4E-BP1), resulting in transcription and translation needed for cell growth, metabolism, and autophagy (13,22,33). The reduced nephrotoxic effects compared to other immunosuppressive drugs have underscored mTOR inhibitors as a more attractive option for transplant recipients (2).
The humoral response is a key component of the immune response against influenza (7,8,18). The immunogenicity of a vaccine is often determined by assessing serum levels of antiflu antibodies, but immunocompromised individuals have been shown to possess lower titers following vaccination compared with healthy controls (6). We, therefore, investigated whether daily treatment of mice with rapamycin compromises induction of protective humoral immune response to a primary lethal H1N1 influenza infection and a subsequent heterosubtypic H3N2 virus challenge.
Materials and Methods
Mice, treatment, and viral infection
Six-week-old C57BL/6 mice (Jackson Laboratories) were treated intraperitoneally (i.p.) daily (d) for the duration of the experiment with 500 μL of phosphate-buffered saline (PBS) containing 12 μg (high dose) or 1.5 μg (low dose) rapamycin (Rapamune oral solution, 1 mg/mL sirolimus; Pfizer) as described previously (1,11,17). Three days after treatment was initiated, mice were inoculated intranasally (i.n.) with 1.5 LD50 of A/Puerto Rico/8/1934 (A/PR8) (H1N1) under avertin anesthesia (Supplementary Fig. S1A; Supplementary Data are available online at
Lung collection and analysis
As described previously (28), lungs were collected, frozen (−80°C), homogenized, and clarified before serial titration in 10- to 11-day-old embryonated hen eggs. The inoculated eggs were incubated at 37°C for 48 h. Allantoic fluid was collected and titrated by hemagglutination assay and viral titers were expressed as the 50% egg infectious dose (EID50).
Serum collection and analysis
Blood was collected in SST Microtainer tubes (Becton, Dickinson & Company) using mandibular bleeds. Serum was isolated by centrifugation and frozen at −40°C until testing. Serum was treated with Receptor Destroying Enzyme (Denka Seiken Company) at 1:4 ratio and incubated for 18 h at 37°C and at 56°C for 30 min.
Virus-specific ELISA was performed by coating flat-bottom immuno 96-well plates (Fisher) overnight with 50 hemagglutinin units (HAU) of homologous virus in 50 μL per well. After blocking with 4% BSA (Sigma-Aldrich) in 1× PBS/0.05% Tween (PBST) (Gibco, Life Technologies), serum was serial diluted twofold or fourfold from 1:100 dilution and secondary HRP-conjugated anti-IgM or IgG (Southern Biotech) was added, respectively. After PBST wash, 3,3′,5,5′-Tetramethylbenzidine (TMB) (eBioscience) and Stop solution (Kirkegaard & Perry) (50 μL) were added at a 1:1 ratio. ELISA background was subtracted, and endpoint titers with optical density (OD) values reaching ≥0.1 were plotted. The limit of detection for ELISA titers was 3.32 log4 units for IgG and 6.64 log2 units for IgM. Hemagglutination inhibition (HI) assay with 1% turkey red blood cells was performed as previously described (27). The limit of detection for HI titers was 2.32 log2 HI units/mL.
Intracellular cytokine assay
On day 35 (5 weeks) postinfection, single cell suspensions from mouse spleen were prepared in MACS buffer (500 mL PBS, 2.5 mL fetal bovine serum, and 2 mL EDTA) using a cell strainer (VWR). T cells and intracellular cytokine (IFNγ and TNFα) staining after in vitro stimulation of 106 splenocytes/well for 15 h with 0.1 multiplicity of infection of virus were analyzed using Alexa Fluor 700-anti-CD8, PE-Cy7-anti-CD4, PerCP-Cy5.5-anti-IFNγ, and APC-anti-TNFα following the manufacturer's recommendations. Approximately, 105 cells were acquired and analyzed on a BD LSRFortessa flow cytometer (BD Bioscience). Data were analyzed with FlowJo software (Tree Star).
Western blot
A549 cells were treated overnight with two different batches of Rapamycin at equivalent dose per volume used within mice weighing 20 g. P70 S6 Kinase phosphorylation (Thr389) was induced using Actinomycin D (Sigma-Aldrich), and anti-p70 S6 phosphorylation-specific 108D2 antibody (108D2; Cell Signaling) was used for detection, as described previously (19).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 software (GraphPad Software). Comparisons were made among infected animals undergoing different treatments (indicated on graphs). Nonparametric one-way analysis of variance Kruskal–Wallis test with Dunn's post-test was used for analysis of virus titer, ELISA, and HI assay. The Student t-test was used to analyze flow cytometry. Survival curves were analyzed using Kaplan–Meier test. All differences were considered statistically significant when the p-value was ≤0.05.
Results
To address the effect of high and low dose of daily rapamycin treatments during a lethal influenza virus infection, as shown in Supplementary Figure S1, we treated mice with two concentrations of the drug (12 or 1.5 μg/d in 500 μL PBS). Function of rapamycin was confirmed by assessing phosphorylation resulting from the activation of the mTOR pathway. As described in the Materials and Methods section, Actinomycin D was added to the A549 cell culture to ensure mTOR activation. Phosphorylation of p70 S6 Kinase, downstream of mTOR signaling, was absent with the addition of rapamycin used in the studies herein (Supplementary Fig. S1B). Intraperitoneal rapamycin treatments in mice began 3 days before infection while controls were only given PBS. We inoculated mice i.n. with 1.5 LD50 of A/PR8 virus (H1N1) as a primary infection on day 0, while control groups were administered PBS alone. Each group was subsequently divided into two subgroups, receiving either a heterosubtypic challenge with 5 LD50 A/HK68 virus (H3N2) or PBS (Supplementary Fig. S1A) to assess whether rapamycin treatments interfere with the induction of cross-protective responses against an antigenically distinct virus.
Following A/PR8 inoculation, similar morbidities were observed in animals treated with high and low concentrations of rapamycin compared to infected control animals (Fig. 1A, left panel). A significant (p ≤ 0.01) delay, however, was observed during the onset of morbidity in infected animals treated with a high concentration of rapamycin reaching 12.5% weight loss (the midway point between 100% and 75% weight) ∼2 days after no or low rapamycin treated mice (Fig. 1A, middle panel). Despite this delay, comparable levels of lethality were observed among all mice challenged with A/PR8 virus, regardless of rapamycin dose (Fig. 1A, right panel). Furthermore, viral titers in lungs collected at days 6, 9, and 12 post A/PR8 inoculation were similar among infected-animal groups, with no significant difference (Supplementary Fig. S2). In summary, our data suggest that rapamycin does not exacerbate morbidity, mortality, or increase lung viral titers during a lethal A/PR8 infection, but rather contributes to delays in the onset of morbidity and recovery among infected animals.

Morbidity, mortality, and immune responses postprimary A/PR8 infection with or without rapamycin treatment.
Next, we examined the effect of rapamycin treatment on the induction of A/PR8-specific antibody levels following primary A/PR8 virus infection. We observed no significant differences in HI titers among infected animals with or without rapamycin treatments (Fig. 1B). Similarly, we observed no significant difference in A/PR8-specific IgG or IgM levels among infected animals receiving no, low, or high dose rapamycin treatments in serum collected 2 and 5 weeks postinfection (Fig. 1C). However, compared to A/PR8 infected mice, at day 35 postinfection, ex vivo analysis of cytokine producing T-cell subsets in high dose rapamycin treated A/PR8 infected mice spleen showed a significant increase in IFNγ+ CD4 T cells, TNFα+ CD4T cells, and IFNγ+ CD8 T cells (Fig. 1D).
Heterosubtypic immunity is crucial in conferring protection against an antigenically distinct virus (9,25). To test whether rapamycin treatments interfere with heterosubtypic immunity, we challenged animals 5–6 weeks post A/PR8 infection with 5 LD50 of an H3N2 virus (A/HK68). Mice exposed to the A/PR8 lethal infection, with or without having undergone daily rapamycin treatments, did not exhibit morbidity or mortality following a lethal challenge with A/HK68 virus (Fig. 2A, left and right panels). Finally, serum levels of anti-A/PR8 and anti-A/HK68 antibodies were assessed 2 weeks post A/HK68 challenge. Animals with previous exposure to A/PR8 continued to maintain their elevated A/PR8 HI titers postchallenge (Fig. 2B, top panel). Among A/PR8 inoculated animals postchallenge, a significant increase in A/HK68-specific HI titers was observed in A/HK68-infected mice compared to their nonchallenged counterparts treated with no (p ≤ 0.001), low (p ≤ 0.0001), or high (p ≤ 0.0001) rapamycin (Fig. 2B, bottom panel). These elevated titers, however, were similar among A/HK68-infected animals, previously exposed to A/PR8, compared to each other. Before challenge, no animals showed detectable A/HK68 HI titers (data not shown). These findings indicate that rapamycin treatments did not impede induction of antibody titers after lethal primary infection and subsequent heterosubtypic viral challenge and did not enhance susceptibility to influenza compared to the infected animals not receiving daily rapamycin treatments.
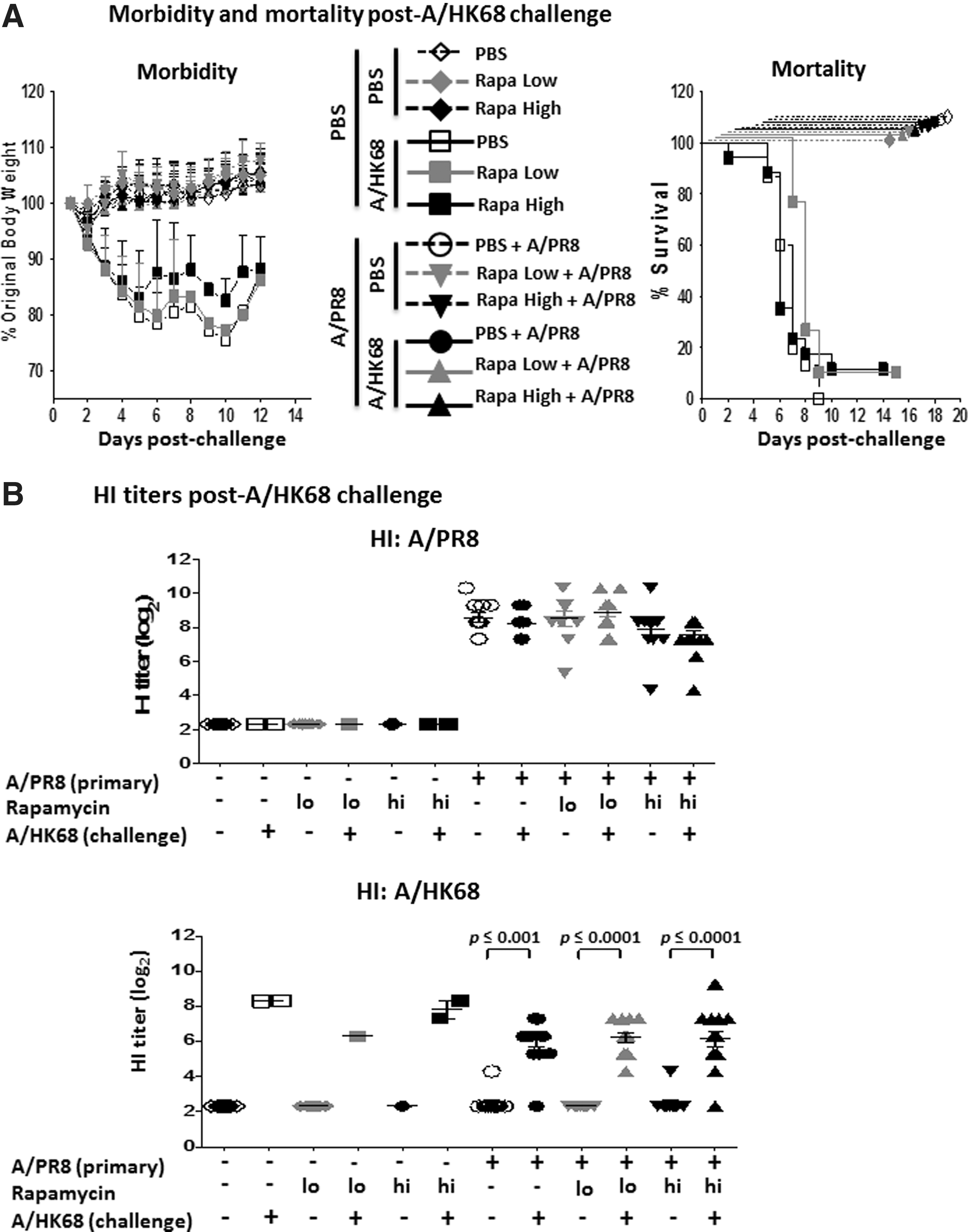
Morbidity, mortality, and HI titer post A/HK68 challenge with or without rapamycin treatment.
Discussion
Medically-induced immunosuppression can compromise an individual's ability to respond to pathogens. In the study herein, we assessed whether rapamycin compromises induction of antibody responses to lethal influenza infections in mice. Our data indicate that rapamycin treatments, despite an increase in cytokine producing T cells, result in similar morbidity, mortality, and antibody responses in A/PR8 and A/HK68-infected animals. Overall, rapamycin did not impede susceptibility or induction of protective antibody responses to lethal H1N1 and H3N2 infections.
Several studies have used low dose (1,11,17) and high dose of rapamycin i.p. treatments (1,24) in experimental animal models and have shown the resulting blood levels to be 5–20 and 40–100 ng/mL, respectively (1). At both high and low dose of rapamycin treatment, we did not observe changes in body weight resulting from these treatments alone. However, among the infected groups, a delay in the onset of morbidity was observed in those treated with the high dose rapamycin. This delay was not reflected in overall morbidity, mortality, or viral clearance among infected animals. Furthermore, HA-specific neutralizing and virus-specific IgG and IgM antibodies were comparable among infected animals despite the daily treatments of rapamycin. Moreover, upon heterosubtypic virus challenge, cross-protective immunity was maintained with both high and low dose daily rapamycin treatments.
Despite its well established application as an immunosuppressant in clinical settings and a suppressor of T-cell proliferation in vitro (5), rapamycin treatment has also been shown to enhance T-cell function in experimental models such as with bacterial (11,24) and viral infections (1). Enhanced T-cell function was also observed in our studies, particularly in mice treated with high dose rapamycin. However, in contrast to findings from Goldberg et al. in which low dose rapamycin treatment led to enhanced pathogen (LCMV, Listeria, and West Nile virus) clearance (15), we did not see any significant differences in lung virus titer/clearance. Although we observed a significant increase in percent influenza (nucleoprotein)-specific CD8+ T cells (data not shown) and T-cell cytokine function in response to rapamycin treatment at day 35 postinfection before heterosubtypic viral challenge, further studies are needed to characterize the kinetics, absolute numbers, and phenotype of CD8 T cells in the secondary lymphoid organs (spleen and draining lymph node) and effector sites such as the lungs. More recently, Keating et al. showed an enhanced protective immunity mediated by modification in virus-specific antibody responses in mice undergoing rapamycin treatments during intraperitoneal priming with influenza virus (17). In this study, the investigators used whole virus for immunization through intraperitoneal route along with one concentration (75 μg/kg) of daily rapamycin treatment. Our data are in agreement with this study's results as morbidity, mortality, and HI titer resulting from lethal infection were unimpeded and extend these findings using two treatment concentrations of rapamycin during the entire course of primary lethal infection and challenge through the natural intranasal route of infection. We did not see lower virus-specific serum IgM antibody levels as reported by Keating et al., which can be due to differences in virus dose, virus subtype, route of virus administration, duration of rapamycin treatment in relation to virus challenge, and time of serum collection postinoculation.
Rapamycin treatment is typically a long-term regimen currently used to prevent graft rejections and may soon be used to treat a variety of cancers and autoimmune diseases (10,23). As a result, these treatments are likely to overlap with influenza seasons. We show that rapamycin treatment neither enhanced the susceptibility to nor compromised induction of virus-specific antibody against influenza infection in a mouse model. While our study focused on the role of rapamycin, patients on long-term immunosuppressive therapy receive a variety of immunomodulatory agents, and therefore, additional studies are warranted to investigate the role of rapamycin in combination with other clinically relevant drugs in the context of influenza virus infection. Future studies investigating the effects of rapamycin in the context of transplantation, autoimmune disease, or cancer on induction of protective immune response to influenza vaccination will allow for a greater understanding of the role this drug may play in “high risk” populations.
Footnotes
Acknowledgments
The authors acknowledge the Atlanta Research and Education Foundation (AREF) (Atlanta VA Medical Center, 1670 Clairmont Road, Decatur, GA 30033). The authors also acknowledge the Association of Public Health Laboratories (APHL) (8515 Georgia Avenue, Suite 700, Silver Spring, MD 20910) for the Emerging Infectious Disease (EID) Fellowship awarded to E.N.J. and the Institutional Research and Academic Career Development Award (IRACDA) for the Fellowships in Research and Science Teaching (FIRST) (Whitehead Biomedical Research Bldg., Suite 648, Atlanta, GA 30323) awarded to J.S.L. The authors thank the animal facilities' technicians, as well as Dr. Samuel Amoah, for their assistance in caring for and treating animals.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.