
Abstract
Plasmacytoid dendritic cells (pDCs) play an important role in innate immune response against viruses, mainly through interferon-α (IFN-α) secretion. Impaired IFN-α secretion has been observed in patients with acute human immunodeficiency virus type 1 (HIV-1) infection and the reasons for this impairment are still obscure. To know the grounds behind this situation, HIV-1 viral copy numbers similar to those found in primary HIV-1 infection were used to stimulate peripheral blood mononuclear cells (PBMCs) and pDCs in this study. Intracellular IFN-α production was seen as early as 2 h in pDCs with TLR-7 agonist (imiquimod) stimulation, but HIV-1 required 48 h to induce secretion of IFN-α in supernatants and it was 10 times less compared to imiquimod. Thus, it shows that HIV-1 delays and impairs IFN-α production from pDCs. Furthermore, the IFN-α inhibitory activity of HIV-1 was checked by stimulating PBMCs and pDCs with imiquimod either simultaneously with HIV-1 or after 2 h pre-exposure to HIV-1. Pre-exposure to HIV-1 resulted in significant reduction in IFN-α secretion by pDCs and PBMCs when compared to imiquimod alone. In addition, simultaneous stimulation of these populations with HIV-1 and imiquimod resulted in significant impairment in IFN-α production in pDCs but not in PBMCs. HIV-1 not only fails to induce IFN-α in adequate quantities but also inhibits IFN-α secretary capacity of pDCs. HIV-1 particles were found to bind CD303 receptor on pDC surface probably blocking initiation of cascade leading to IFN-α impairment. The understanding of the pathways that lead to this suppression may help in devising the HIV control strategies.
Introduction
P
It is known that engagement of endosomally present TLR-7 and 9 by ssRNA and ds CpG DNA, respectively, leads to the production of IFN-α (11). The activation by these TLRs in pDCs results in rapid and robust IFN-α production accounting for 3–10 pg IFN-α per cell within 24 h, which is 100–1,000 times more than any of the other IFN-α producing cell type (18). This robust production of IFN-α is due to the constitutive presence of IRF-7 protein in pDCs, which upon TLR triggering migrates to the nucleus and facilitates IFN-α transcription (12,15,30), bypassing classic autocrine feedback involving IFN-β (8,18).
Decreased pDC numbers and functions have been reported in human immunodeficiency virus type 1 (HIV-1) infection and the low pDC frequencies are found to be associated with high viral load and low CD4+ cell count, resulting in rapid disease progression (3,10,13). The HIV-infected individuals showing slow or no disease progression in the absence of antiretroviral treatment such as long-term nonprogressors maintain high pDC numbers as well as near-normal functionality; on the contrary, HIV-infected patients who show rapid disease progression have low pDC numbers (22). Patients in the acute phase of HIV infection have shown impaired pDC function in terms of decreased IFN-α production, which in turn may be recognized as one of the determinants of the rate of disease progression (4,9,10,14,16,21,32). In the simian immunodeficiency virus (SIV) infection, pDCs have been shown to accumulate at the infection sites early after infection but are unable to control infection even in the presence of proinflammatory cytokines (17). This supports the hypothesis that HIV-1 may also have an inhibitory effect on pDC function, thus evading immune control during early phase of infection. However, the mechanisms underlying this inhibition are still unknown.
Several studies have reported secretion of abundant quantities of IFN-α, upon in vitro stimulation with HIV-1. However, HIV-1 viral copy numbers used for stimulating the cells in these studies were very high (equivalent of 300–900 ng/mL of p24 antigen) (4,21,22) compared to virus copy numbers seen in individuals with early HIV infection (equivalent of 6–7 ng/mL of p24 antigen) (2,6,19,23,31). Hence, in vitro experiments using HIV-1 copy numbers similar to those found in in vivo conditions may reflect the influence of HIV-1 on IFN-α production in primary infection more accurately. Also, we need to know whether such low viral copy numbers of HIV-1 are able to actively inhibit IFN-α production from pDCs or not, considering the possibility that low in vivo doses of HIV-1 may not be able to stimulate IFN-α production in pDCs during primary HIV-1 infection. We report here that HIV-1 at doses similar to in vivo conditions found in recent HIV-1 infection not only fails to stimulate IFN-α production in pDCs but also actively downregulates the IFN-α production.
Materials and Methods
Virus stock
The HIV-1 C strain was isolated from recent seroconverter peripheral blood mononuclear cells (PBMCs), which were available in the institute. Virus isolation was performed according to the methodology described earlier (20). Briefly, PBMCs purified from peripheral blood of HIV-infected person were cocultivated with phytohemagglutinin P-stimulated (48 h) PBMCs from HIV-1-uninfected individuals for 21 days. Culture supernatants were periodically checked for the presence of HIV-1 p24 antigen (HIV-1 p24 Antigen Capture Assay; ABL, Inc., Rockville, MD). After appearance of p24 antigen in culture supernatant, titer of virus was estimated by TCID50 assay with the Spearman–Karber method of titer calculation. Virus was then stored at −80°C.
Viral copy estimation
HIV-1 viral copies present in 0.01 or 0.5 multiplicity of infection (MOI) viral inoculums were quantified using the Roche Cobas® AMPLICOR HIV-1 MONITOR Test, V1.5 (Roche Diagnostics, Basal, Switzerland).
PBMC preparation and stimulation with HIV or imiquimod
PBMCs from peripheral blood or buffy coats collected from healthy donors were separated by density gradient centrifugation using Ficoll/Hypaque (Sigma Aldrich, St. Louis, MO). PBMCs were plated as 1 × 106 cells per milliliter per well in 24-well plate in RPMI 1640 with 10% fetal bovine serum (FBS; Moregate Biotech, Bulimba QLD, Australia), L-glutamine (2 mM) (Gibco-Invitrogen, Carlsbad, CA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco-Invitrogen) and stimulated with 20 μg/mL imiquimod (TLR-7 agonist) (R&D, Minneapolis, MN) or 0.01 MOI of primary HIV-1 isolate for specified time periods. Imiquimod was used as positive control because of its ability to induce IFN-α through TLR-7.
pDC preparation and stimulation with HIV or imiquimod
pDCs were isolated from PBMCs of healthy donors or buffy coats, by depletion of non-pDCs using the pDC isolation kit by following the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). Purity of enriched pDCs ranged between 85% and 99% as measured by the presence of CD123 and CD303 (BDCA-2) marker on flow cytometer (BD FACS Aria, San Jose, CA).
Purified pDCs were plated as 2 × 104 cells per well in 96-well plate per 100 μL RPMI 1640 with 10% FBS (Moregate Biotech), L-glutamine (2 mM), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco-Invitrogen) supplemented with 20 ng/mL IL-3 (e-Biosciences, San Diego, CA) and left overnight at 37°C in 5% CO2 atmosphere. pDCs were stimulated the next day with 20 μg/mL imiquimod or 0.5 MOI HIV-1 primary isolate.
Frequencies of cells secreting IFN-α in response to imiquimod or to HIV-1 as measured by intracellular cytokine staining assay
PBMCs were stimulated for 2, 4, or 6 h with imiquimod (20 μg/mL) or left unstimulated as cell control wells, while pure pDCs were stimulated for 8 h with imiquimod (20 μg/mL) or 0.1, 0.5, or 1 MOI of HIV-1 primary isolate or left unstimulated. Brefeldin A-10 μg/mL (Sigma Aldrich) was added after an hour of initiation of incubation periods to inhibit protein transport. Following stimulation for specified time periods, 20 mM EDTA was added to the wells and plate kept overnight at 4°C. On the next day cells were stained for surface markers with lin 1-FITC, HLA-DR-Per CP, CD11C-PE, CD123-PECy7 (all from BD Biosciences, San Jose, CA) for PBMCs and CD123-PECy7 (BD Biosciences) and CD303 (BDCA-2)-FITC (Miltenyi Biotec) for pDCs for 30 min. Cells were then fixed and permeabilized with the fixation and permeabilization solution (BD Biosciences). Intracellular IFN-α was detected with the IFN-α-Alexa Fluor 647 (BD Biosciences) antibody incubated for 1 h at room temperature. 103 pDCs were identified as lin1 and CD11C negative and HLA-DR+CD123+ from PBMC culture and CD123+CD303+ cells from pure pDC culture on flow cytometer (BD FACS Aria). Percentages of pDCs with intracellular IFN-α were analyzed using FACS Diva software.
IFN-α secretion by pDCs in response to imiquimod with or without pre-exposure to HIV-1 as measured by ELISA
Purified pDCs were stimulated with 20 μg/mL imiquimod for 8 h or with 0.5 MOI HIV-1 primary isolate for 24 and 48 h. Whereas the PBMCs and pure pDCs were stimulated with imiquimod alone (20 μg/mL) or HIV-1 alone or with imiquimod (20 μg/mL)+HIV-1primary isolate (0.01 MOI for PBMCs and 0.5 MOI for pDCs) for 8 h or initially with HIV-1 primary isolate for 2 h and then with imiquimod (20 μg/mL) for 8 h. Supernatants were collected after pelleting the cells and stored at −20°C. IFN-α levels were quantified by a pan-specific (detects IFN-α subtypes 1/13, 2,3,4,5,6, 7, 8,10,14, 16, and 17) IFN-α ELISA (Mabtech, Nacka Strand, Sweden) according to the manufacturer's instructions.
Measuring attachment of CD303 antibody to pDCs with or without HIV-1 exposure
Pure pDCs from healthy donor were plated as 2 × 104 cells per well per 100 μL RPMI with 10% FBS in 96-well U bottom plate and exposed to 5, 10, 15, or 20 MOI of HIV-1 or left unexposed for 1 h at 37°C. Then fixed with fix buffer–I (BD Biosciences) and stained with CD123-PE (BD Biosciences) and CD303-FITC (Miltenyi Biotec) antibody. The samples were then acquired on FACS Aria (BD Biosciences) and analyzed using FlowJo software.
Ethics statements
The study was approved by the Institutional Ethics Committee of National AIDS Research Institute (Pune, India). Blood samples were collected from HIV-negative donors after taking their written informed consent or buffy coats were obtained from a local blood bank.
Results
Imiquimod stimulates intracellular IFN-α in pDCs as early as at 2 h
First, we wanted to determine the minimum time required for IFN-α production after stimulation with positive control; PBMCs from healthy HIV-negative individuals were stimulated with 20 μg/mL of imiquimod for 2, 4, or 6 h and then fixed, permeabilized, and stained as described in the Materials and Methods section. It was observed that in pDCs, intracellular IFN-α production starts as early as 2 h after imiquimod stimulation (10.56% pDCs secrete IFN-α at 2 h) and plateaus between 4 h (25.66% pDCs secrete IFN-α) and 6 h (27.44% pDCs secrete IFN-α) (Fig. 1).

Imiquimod stimulates intracellular IFN-α in pDCs as early as at 2 h.
Exposure to HIV-1 does not stimulate pDCs to produce IFN-α
2 × 104 pure pDCs were stimulated with either imiquimod (20 μg/mL) or with HIV-1 (0.1 MOI: 6.168 × 107 viral copies or 0.5 MOI: 3.24 × 108 viral copies or 1 MOI 6.4 × 108 viral copies) or kept unstimulated as control for 8 h. Intracellular IFN-α production was observed by flow cytometry. 19.78% pDCs exhibited intracellular IFN-α after 8 h of imiquimod stimulation, but minimal (0.5%) or no IFN-α was observed after stimulation with HIV-1 at 8 h (Fig. 2).
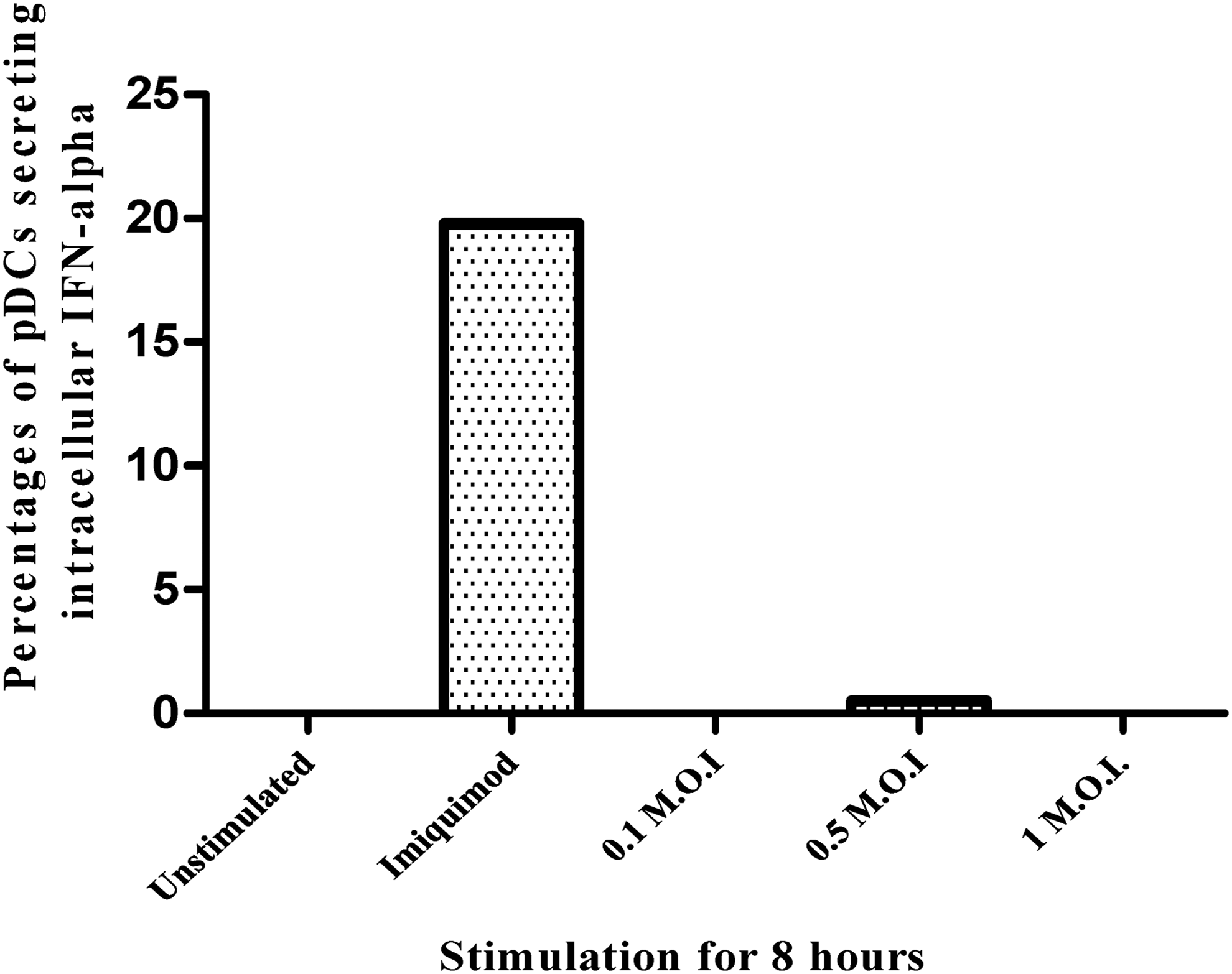
Exposure to HIV-1 does not stimulate pDCs to secrete IFN-α. Mean percentages of pDCs secreting IFN-α from four healthy donors when pure pDCs were stimulated with imiquimod (20 μg/mL) or with HIV-1 primary isolate-0.1 MOI or 0.5 MOI or 1 MOI or with media control for 8 h. HIV-1, human immunodeficiency virus type 1; MOI, multiplicity of infection.
Exposure to HIV causes delayed and lower IFN-α secretion
2 × 104 pure pDCs were stimulated with 0.5 MOI HIV-1 or with TLR-7 agonist, and IFN-α in the supernatants was measured using pan-specific ELISA at 24 and 48 h for HIV-1, while at 8 h for TLR-7 agonist. No IFN-α production was seen in HIV-treated wells at 24 h, however, at 48 h, minimal IFN-α secretion (561 pg/mL) was observed, which was 10 times lower than the IFN-α secretion seen in imiquimod-treated wells (5,020 pg/mL) (Fig. 3).
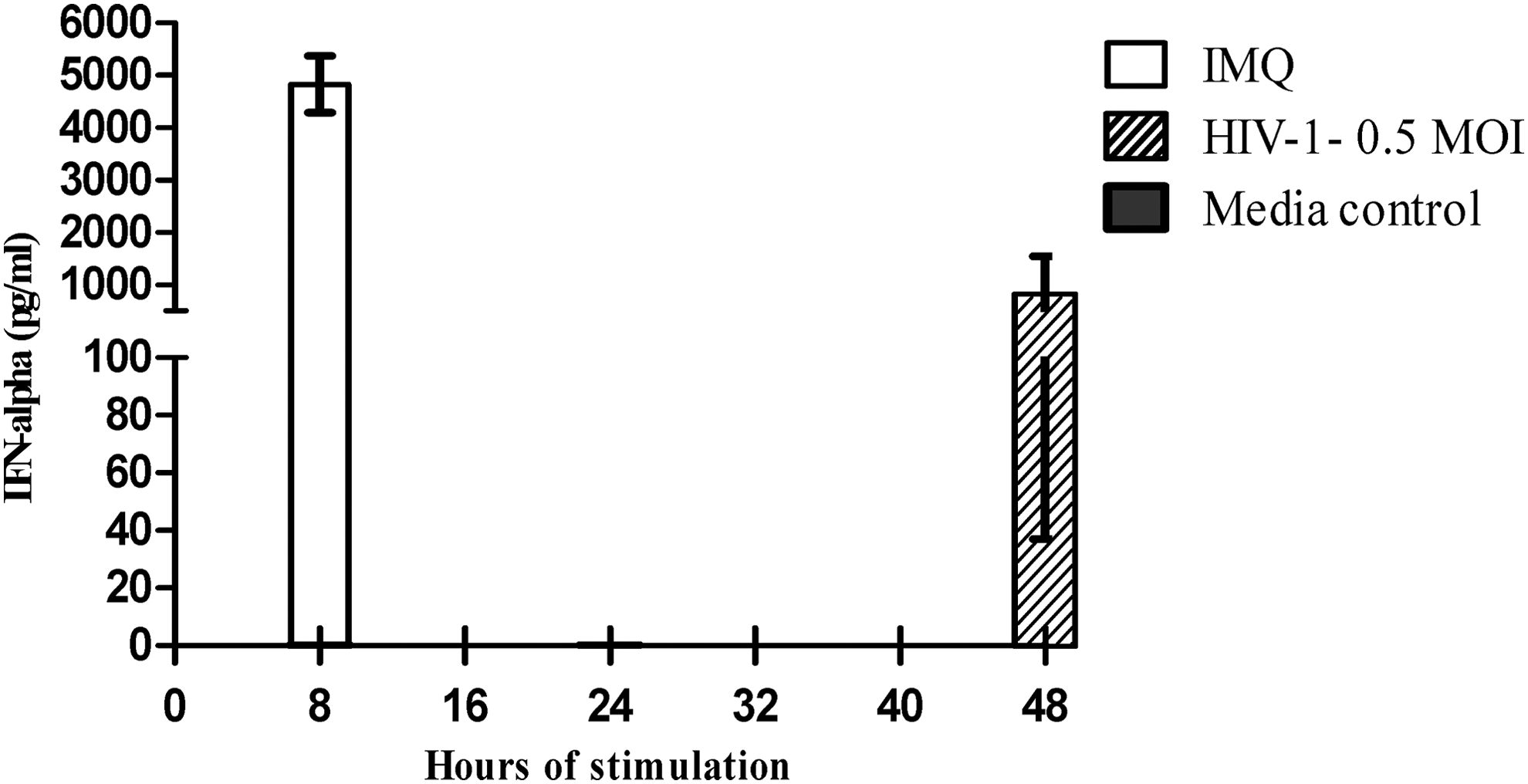
Exposure to HIV delays and suppresses IFN-α production. Y-axis denotes IFN-α produced at each tested time period after pure pDCs were stimulated with imiquimod (IMQ) for 8 h or with HIV-1 for 24 or 48 h. Shown here are the average values of IFN-α from four healthy individuals measured by ELISA (pan specific).
Exposure to in vivo doses of HIV-1 inhibits IFN-α production induced with imiquimod
To check whether the lower production of IFN-α exhibited by pDCs upon HIV-1 exposure is due to lower activation levels by low copy numbers of virus or HIV-1 actively suppresses IFN-α secretion by pDCs; PBMCs were exposed to in vivo doses of HIV-1, that is, 0.01 MOI (3.24 × 108 viral copies) with or without 20 μg/mL imiquimod. In addition, one million PBMCs were pre-exposed for 2 h to 0.01 MOI of HIV-1 followed by treatment with 20 μg/mL imiquimod. Appropriate media and unstimulated cell controls were included. As assessed by ELISA, the media control and in vivo dose of HIV-1 did not show IFN-α secretion from PBMCs. When cells were exposed to HIV-1 and imiquimod simultaneously, IFN-α production was not inhibited. However, when PBMCs were preincubated with HIV-1 for 2 h and then stimulated with imiquimod, a significant decrease in IFN-α secretion was noted when compared to imiquimod (191 pg/mL against 1,019 pg/mL) stimulation alone (Fig. 4A).

Exposure to in vivo doses of HIV-1 inhibits IFN-α production induced with imiquimod.
When similar experiments were carried out using purified pDCs, it was observed that exposure to in vivo doses of HIV-1, that is, 0.5 MOI (3.24 × 108 viral copies) resulted in a significant decrease in IFN-α production not only when the cells were incubated with HIV-1, 2 h before imiquimod stimulation (535 pg/mL) but also with simultaneous exposure of imiquimod and HIV-1(2,149 pg/mL) compared to imiquimod alone (6,315 pg/mL) (Fig. 4B).
HIV blocks the binding of antibodies to CD303 on pDCs
The binding CD303 receptor has been shown to be associated with abrogation of IFN-α production in pDCs. One of the mechanisms by which HIV-1 could suppress IFN-α production by pDCs could be engaging CD303 on the surface of pDCs. Hence, to explore whether the IFN-α inhibitory effect exerted by HIV-1 was linked to HIV binding to CD303 or not, we allowed increasing MOI of HIV-1 to bind to pDCs for an hour and then fixed and stained with CD303 antibody. Figure 5 demonstrates that mean fluorescence intensity (MFI) of CD303-FITC antibody attached to pDCs decreases as MOI of HIV-1 used for pDC exposure increases.
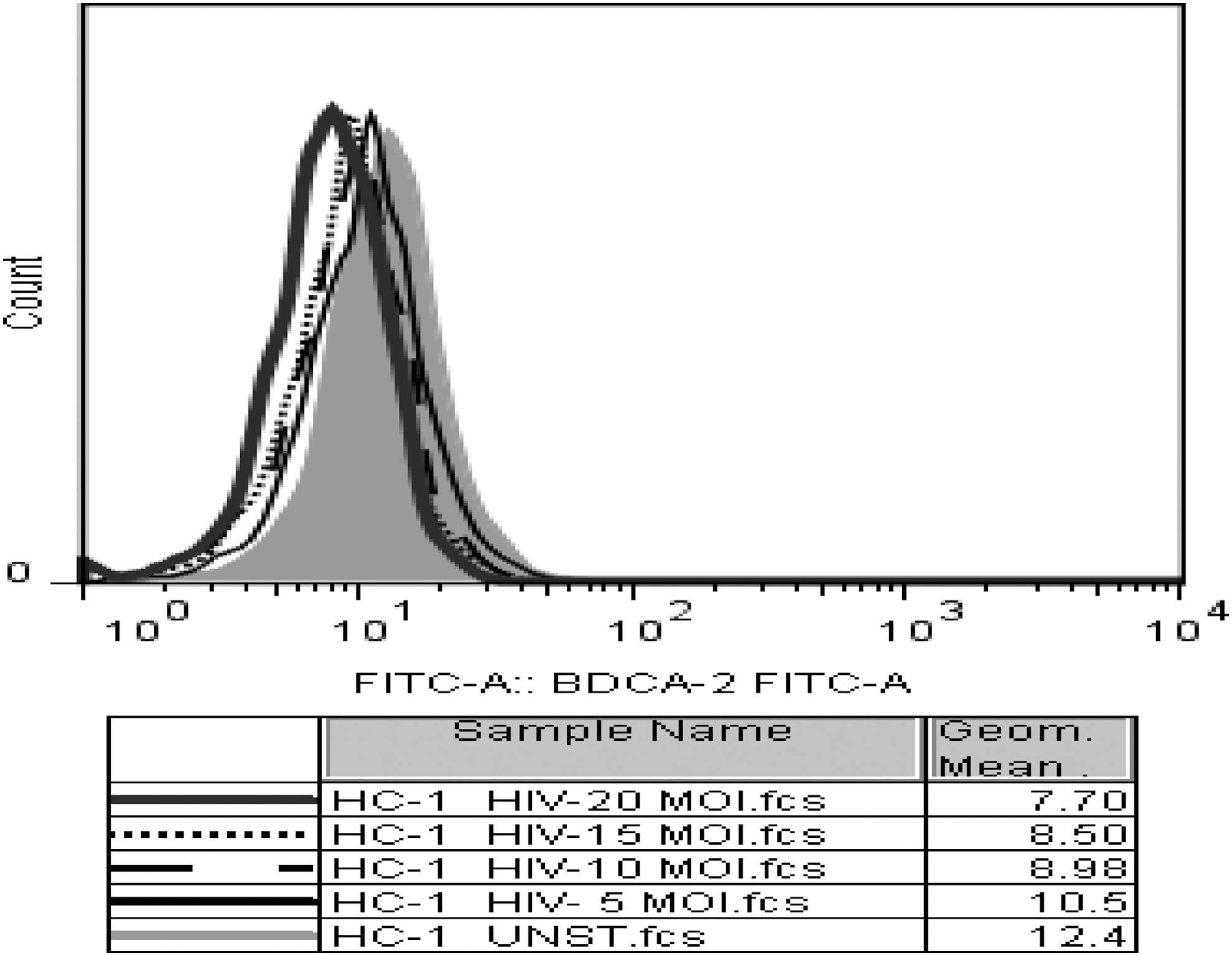
HIV blocks the binding of antibodies to CD303 on pDCs. Pure pDCs were exposed to 5, 10, 15, and 20 MOI of HIV-1 virions or left unexposed followed by fixation and staining with CD303 antibody as described in the Materials and Methods section. The histogram represents MFI of CD303 antibody attached to pDC surface after exposure to HIV-1. The accompanying table shows geometric mean of FITC fluorescence for BDCA-2 antibody. MFI, mean fluorescence intensity.
Discussion
Exposure to HIV-1 copy numbers higher than those seen in the acute HIV infection results in the robust induction of IFN-α in vitro (4). We report here that when virus copies at numbers similar to the actual transmission setting were used to stimulate pDCs, there was lower as well as delayed production of IFN-α when compared with IFN-α production by TLR-7 agonist; imiquimod and we also showed that the HIV-1-exposed pDCs could not secrete IFN-α even when they were stimulated with TLR-7 agonists afterward. This suggests that decreased or delayed IFN-α production from pDCs is not a result of lower stimulation with HIV-1, but exposure to HIV-1 itself inhibits IFN-α production through mechanisms not yet identified.
IFN-α levels have been found to be low in peripheral blood from patients with primary HIV-1 infection (24), but the mechanisms causing these lower levels are still unknown. Various in vitro studies have documented robust IFN-α production in supernatants of PBMCs activated by HIV-1. However, the viral copy numbers used for this activation were 20- to 100-fold higher than those found at early infection sites (4). Lo et al. have demonstrated that in vivo doses of HIV-1 cause lower level of IFN-α production (19). Hence, in an attempt to replicate scenario at the time of HIV transmission, we used 108 viral copies for stimulation of pDCs in our experiments, which are similar to the one used in in vivo sexual transmission studies in rhesus monkeys and normally found in blood of recently HIV-1-infected persons (6,25). Our study showed that HIV-1 doses reflecting in vivo conditions of acute HIV-1 infection resulted in late and impaired secretion of IFN-α production by pDCs when compared to imiquimod stimulation. We observed that IFN-α production induced by imiquimod starts as early as 2 h and peaks at 4 h at levels much higher than induced by HIV-1 stimulation. These results are consistent with earlier findings reported by Lo et al., where they have compared kinetics of IFN-α production on stimulation/infection by HIV-1 with other viruses such as influenza, Sendai, and herpes simplex virus-2 (HSV-2) (19).
To study whether HIV-1 actively suppresses IFN-α production in PBMC and pure pDC cultures, we stimulated cells with imiquimod simultaneously or after pre-exposure to HIV-1 at in vivo doses. No suppression of IFN-α production was observed when PBMCs were simultaneously stimulated with imiquimod and HIV-1, but when pure pDCs were stimulated with imiquimod and HIV-1 simultaneously there was a significant decrease seen in the amount of IFN-α produced (Fig. 4). PBMCs include cell types other than pDCs that might have contributed to IFN-α levels upon exposure to HIV-1 in our study. In addition, simultaneous addition of imiquimod, along with HIV-1, may have resulted in immediate IFN-α secretion from pDCs but at lower levels due to HIV-1-mediated inhibition. Thus, IFN-α produced by pDC and non-pDC types in PBMC culture may have contributed to IFN-α at levels comparable to imiquimod stimulation. Upon prestimulation of pure pDCs and PBMCs with HIV-1 for 2 h, IFN-α response to imiquimod stimulation was suppressed significantly; 12 and 5 times reduction for pure pDCs and PBMCs, respectively, when compared to imiquimod stimulation alone (Fig. 4). This suggests that HIV-1 actively inhibits IFN-α production by pDCs even when they are treated with TLR-7 agonist. Suppression of IFN-α production upon pre-exposure to HIV-1 by PBMCs probably indicates that during exposure time, HIV-1 inhibits IFN-α from pDCs leading to suppressed IFN-α production. IFN-α production from cell types other than pDCs may also be actively influenced by HIV-1 causing added inhibition of IFN-α production. However, as pDCs are the major producer of IFN-α, the inhibitory effect exerted on other cell types may have minimal contribution in IFN-α inhibition observed in HIV-1 pre-exposed PBMC culture.
The IFN-α inhibitory activity of HIV-1 in pDCs can be linked with abrogation of CD303 (BDCA-2) downstream pathway (19). The pDCs demonstrate inhibition of maturation and reduced IFN production after ligating and crosslinking CD303 receptor with anti-CD303 antibody (5,29), and gp120 of HIV-1 has been shown to alter IFN-α production from pDCs by binding to CD303 on pDCs (23). In hepatitis-B infection also HBsAg has been shown to ligate with CD303 signifying a possible role of CD303 interaction in pDC unresponsiveness (33). Hence, we suspect that HIV-1 may be inhibiting IFN-α production from pDCs by attachment to CD303 receptor to alter the CD303 downstream pathway. To see whether HIV-1 virions attach to CD303 receptor on pDCs or not, we incubated pDCs with increasing doses of HIV-1 particles before staining pDCs with anti-CD303 antibody. It was observed that binding of CD303 antibody was suppressed in a dose-dependent manner with increasing doses of HIV-1 particles. The reduction in MFI was directly proportional to infectious doses of virus used for exposure to pDCs (Fig. 5). This suggests that HIV-1 binds to CD303 receptor and obstructs CD303-FITC antibody binding. This finding is consistent with recent studies of Lo et al. (19), where binding of HIV-1 to CD303 receptor has been shown by activation of CD303 downstream pathway by demonstrating phosphorylation of SYK molecule after HIV-1 exposure. Further studies are needed to find out whether other phosphorylation molecules or mechanisms downstream of CD303 pathway could be hijacked by HIV-1 to affect IFN-α production. This will provide insights into how HIV-1 alters typical pDC responsiveness.
The majority of HIV-1 infections are acquired through mucosal route (6) and after entry into these surfaces, various immune cells such as macrophages, NK cells, and pDCs get recruited at the site of infection (28). IFN-α is a very important cytokine at mucosal surfaces for early control and evasion of viruses, as it can produce antiviral state in neighboring cells as well as can activate helper and cytotoxic T cells (28). Our study showed delayed and compromised IFN-α production by pDCs after HIV-1 exposure at doses similar to the one observed in patients with acute HIV-1 infection. Similar suppression of IFN-α production at the mucosal surfaces of an individual is likely to enhance the chances of establishment of HIV infection through subversion of IFN-α production. As HIV-1 replication kinetics are very rapid, the suppressed and late IFN-α production may allow the virus to surpass various antiviral controls mediated through IFN-α, thus leading to establishment and spread of its infection to various pockets of body. Also, as shown in this study, pDCs once exposed to HIV-1 cannot be restimulated by TLR-7 agonist implies that at mucosal surfaces, after interaction with HIV-1, pDCs may be become refractory to other stimuli such as bacteria at early infection phase only, thus making host susceptible to secondary or opportunistic infections. Our findings are contrary to the observations by O'Brien et al. who have demonstrated that pDCs prestimulated with HIV can continue to produce IFN-α and cytokines upon restimulation with HIV or other viruses (26). More studies on pDC activation ex vivo from recently infected individuals may help to delineate pDC and HIV-1 interactions during acute HIV-1 infection.
Footnotes
Acknowledgments
We are thankful toward all healthy donors for donating their blood and the Sahyadri Blood bank, Deccan Gymkhana, Pune, for providing buffy coats during this study. We also express our gratitude toward the Indian Council of Medical Research (ICMR), Delhi, and intramural funding of National AIDS Research Institute, Pune, India, for giving support to this study. Mrs. Dhamanage is ICMR Senior Research Fellow and has been provided a 3-year research fellowship.
Author Disclosure Statement
No competing financial interests exist.