
Abstract
Viruses are the intracellular pathogens that reproduce only in the living cell and manipulate the cellular machinery to produce more viruses. Viral replications can affect cellular genes of the host in multiple cancerous ways. Approximately, 20% of all human oncogenesis is caused by cancer-causing viruses known as oncoviruses. Viral infection causes chronic inflammation leading to cell death, uncontrollable proliferation, and modulated expression of some of the regulatory proteins. Oncogenesis is a multistep phenomenon in which normal host cells are transformed into cancerous cells on the basis of host genetic variability. Oncogenic viruses encode genes that cause viral replication and transformation of the host cells to produce viral proteins and protein complexes. The phenomenon from basic viral infection to tumorigenesis is lengthy due to the involvement of factors like immunity complications, cellular mutations, and exposure to other cancerous agents. The viruses that are involved in human cancer development are Hepatitis B virus (HBV), Hepatitis C virus (HCV), Epstein–Barr virus (EBV), Human papilloma virus (HPV), Kaposi's sarcoma herpes virus (KSHV), and Human T lymphotrophic virus 1 (HTLV-1). This review article summarizes advanced knowledge related to human oncogenic viruses and the molecular mechanisms that lead to tumorigenesis in humans.
Introduction
O
Viral oncogenes play a critical role in uncontrollable proliferations of host cells and synthesize new viral gene-associated proteins that lead to transformation (32). Proto-oncogenes are cellular counterparts of viral oncogenes that are converted to an oncogenic state by mutations, amplifications, deletion, or chromosomal translocations by viral oncogenes as shown in Table 1 (4).
EBV, Epstein–Barr virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HTLV-1, human T lymphotrophic virus; HPV, human papilloma virus; KSHV, karposi's sarcoma herpes virus; MCV, merkel cell polyomavirus.
Oncogenes persistently struggle with the tumor suppressor genes (that protect DNA and control cellular activities), but in case of cancer development, tumor suppressor genes lose their struggle or viral oncogenes prevail by causing inactivation of tumor suppressor genes (68). Thus the viruses itself are not the direct cause of human cancer; oncogenesis occurs after long-lasting chronic infections leading to the development of cancer. Viral interactions with the human immune system and ultimate immune suppression are the reasons of oncogenic initiation (73).
General Mechanisms of Viral Oncogenes
Cancer development is a complicated multistep process that involves complex events to transform a normal cell to a cancerous cell (1). Majority of the tumor viruses in humans encode oncogenic proteins that specifically target cellular proteins, for example, p53 (a tumor suppressor gene that controls cell cycle progression and apoptosis) and retinoblastoma (pRb), which play a significant role in the loss of tumor suppression that leads to cancer development (36,55). Viral oncogenesis is caused due to the presence of viral oncogenes (v-onc), activation of cellular proto-oncogenes, cellular transformations, deregulated cell cycle, and inactivation of tumor suppressors (16).
Presence of viral oncogenes
Viral oncogenes are derived from cellular proto-oncogenes. Products of oncogenes can be categorized as the growth factors, growth factor receptors, transcription factors, signal transducers, and apoptosis regulators (13). Both viral and cellular oncogenes play significant roles in cancer development or cellular oncogenes alone may develop cancer. It is now well established that all human oncogenes and viral oncogenes target human proteins and genomes for the process of cancer development (30,42).
Cellular transformations
Tumor viruses integrate their genetic material into the host cell genome, which is essential for cellular transformations, causing mutations, chromosomal rearrangements, and uncontrolled cell divisions by interfering with the mitogenic signaling pathway and cell cycle processes (54). The transformed cell further leads to an immortal state that grows indefinitely. In DNA viruses, genetic material is directly inserted into the host genome causing neoplasia, while RNA viruses have to reverse transcribe RNA to DNA before insertion into the host genome. Thus, the major cause of host cellular transformation is disrupted regulation of cellular metabolism due to insertion of viral genome into the host cellular genome (54).
Cell cycle deregulation
Cell cycle regulates accurate DNA replication and chromosomal segregation. Cyclins, cyclin-dependent kinases (CDKs), and their inhibitors regulate cell cycle and homeostasis mechanism. Apoptosis is a regulatory procedure for the homeostatic balance of human body and its deregulation leads to unlimited proliferation of transformed cells. It is noted that molecular alterations of host genome by oncoviruses lead to deregulated apoptotic processes and disrupted homeostasis (19). Viruses have evolved many strategies to overcome the regulatory system of cell cycle leading to continuous proliferation of the infected cells. Viral oncogenes increase or decrease the effects of specific cell cycle regulatory proteins and arrest the mutated cells in the synthetic phase of cell cycle that leads to the induction of transcriptional phosphorylation and DNA replication (18).
Inactivation of tumor suppressors
Tumor suppressor genes protect the cells from malignant transformations by instructing the cells to prevent cell growth and division. Viral oncoproteins interfere with tumor suppressor genes function causing deregulated cell growth and uncontrolled cell proliferation. The p53 plays a major role in eliminating or inhibiting abnormal cell proliferation. Hepatitis B (HBV)-encoded hepatitis B X-antigen (HBx) oncoprotein inactivates p53 and blocks p53-mediated apoptosis. Hepatitis C (HCV) containing nonstructural protein 5A (NS5A) interferes with the DNA binding activity of p53 (7). The pRb is a negative regulatory protein of cell cycle. The E7 oncoprotein of human papilloma virus (HPV) interferes with its binding to E2F transcription factor. Thus E7 causes several biological effects like increased transcription, autophagy, and inhibition of interferon signaling (50).
Cancer-causing viruses have got a significant role in the advancement of research related to cancer biology. Ontogenesis includes various hallmarks such as continued cellular proliferation, deregulation of cellular energetics, genetic instability, induction of angiogenesis, immune invasion, and metastasis as summarized in Figure 1 (14).
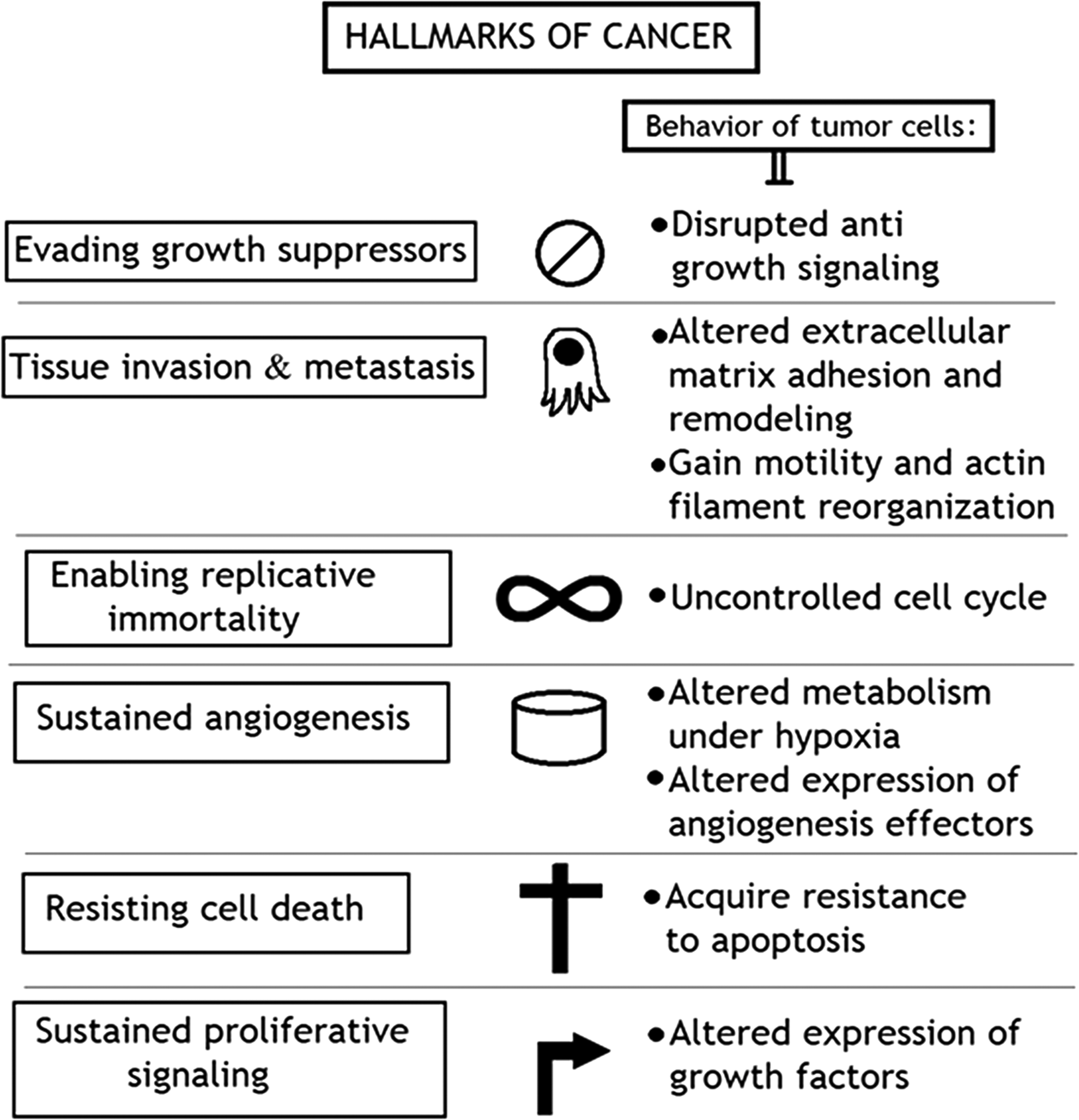
Hallmarks of cancer development.
DNA Oncoviruses and Their Targets
DNA tumor-generating viruses include EBV, HPV, Kaposi's sarcoma herpes virus (KSHV), HBV, and MCV. These oncogenic viruses act in two ways; in permissive cells, viral replication causes cell lysis and death, while in nonpermissive cells, viral DNA is integrated into the host genome. The integrated DNA encodes binding proteins to arrest normal cell growth by acting on regulatory proteins; p53 and pRb. Cellular proteins p53 and pRb are the central targets of oncogenic viruses. Thus, the host cell is transformed to express proteins that facilitate both viral and cellular DNA synthesis (14,32). Tumor necrosis-associated factors (TRAFs) and Nuclear Factor kappa-light-chain-enhancer of activated B cells (NK-κB) are the main target factors for viral oncogenes (5). Hypophosphorylated Rb negatively regulates G1 phase of cell cycle to S phase (synthesis phase) and further blocks the E2F activity (transcriptional factor of eukaryotes), a transcriptional factor that is involved in DNA replication (56). Viral oncoproteins bind specifically to Rb and inactivate its hypophosphorylated form leading to the production of free E2 that further causes uncontrolled proliferation of transformed cells. Latency-associated nuclear antigen (LANA) plays a significant role in KSHV-associated oncogenesis (41).
Human papilloma viruses
An Italian Physician in 1842 noticed that cervical cancer is associated with sexual contact (22). It was suspected that it may have contagious etiology, and in 1907, the transmission of genital warts using cell-free extracts was observed by Ciuffo (10). HPV is a circular, nonenveloped double-stranded DNA virus. HPVs infect mucosal epithelial cells causing cervical cancers. Till today, there are 130 types of HPV discovered. Depending upon the nature of infectivity, HPVs are classified into low-risk and high-risk groups (73). HPV infections are mostly asymptomatic and in almost 90% cases, the infection is spontaneously cleared after 1 or 2 years of infection. However, HPV infection is the most common sexually transmitted disease. There are estimated 0.5 million annual cases of cancer due to HPV infection. It is responsible for the development of warts on cervix, anus, vulva, penis, head and neck area, and oropharynx (46). It is shown that the high-risk tumor generating activities of HPVs, including HPV-16 and HPV-18, give rise to cervical carcinomas where E6 and E7 proteins play primary role in promoting instability of human genome. The low-risk HPV types, including HPV-6 and HPV-11, affect the mucosa of anogenital tract to provoke epithelial hyperplasia (44). E6 and E7 genes of HPVs are the specific causative agents in human tumorigenesis as shown in Figure 2.
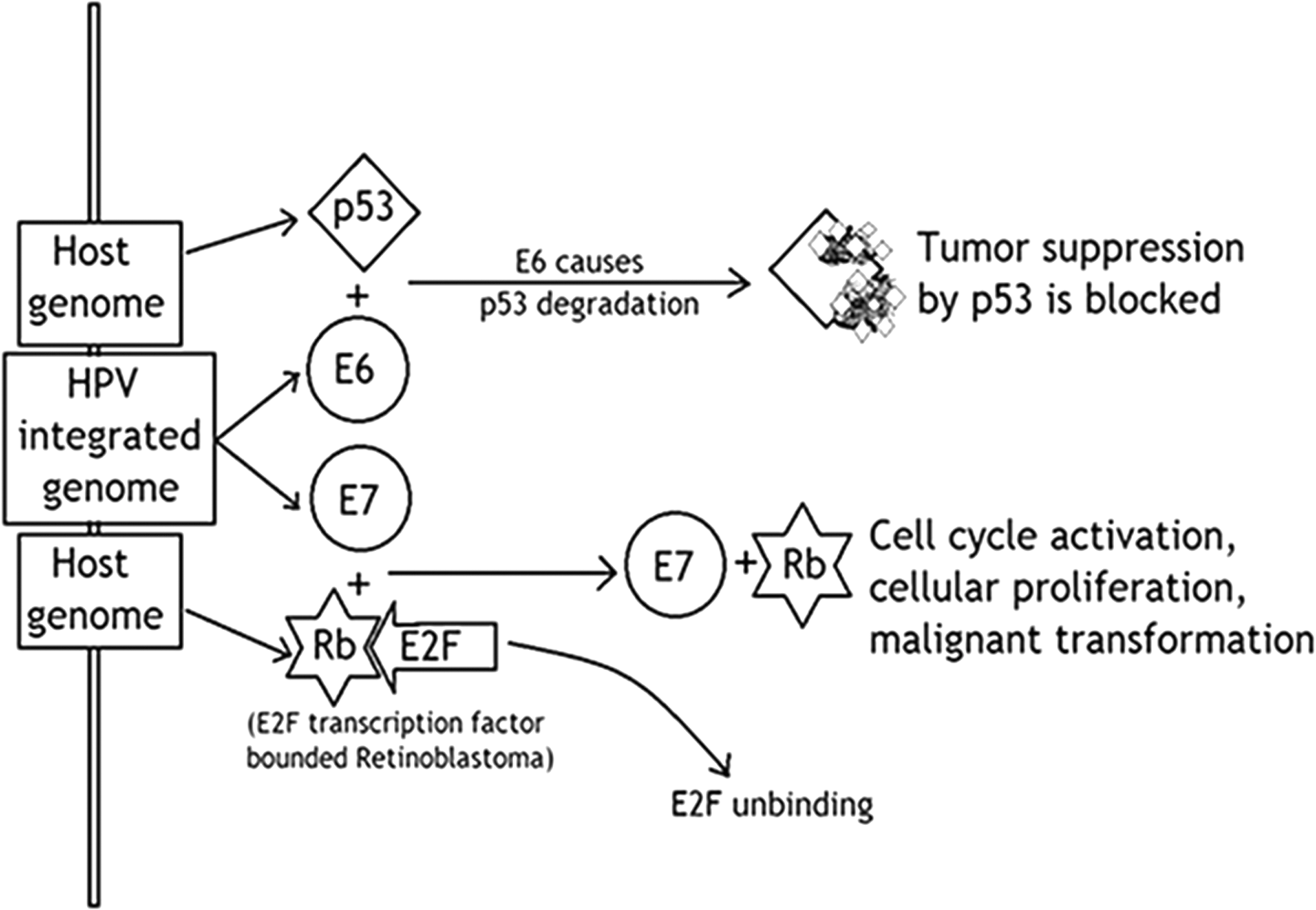
HPV induced oncogenesis: HPV oncogenes E6 and E7 deregulate host cell cycle by binding and inactivating tumor suppressor genes p53 and pRb. Binding of E6 gene to p53 causes degradation of p53. E7 gene binds to pRb and disrupts the complex between pRb and E2F leading to uncontrolled proliferation. HPV, human papilloma virus.
In general, HPV is the most dominating oncovirus among all oncoviruses. Approximately, 5% of all human cancers are caused by HPV infections (28). Oncogenic behavior of HPV is associated with several selected gene mutations, including p53, IL-10, retinoic acid receptor (RAR), and human leukocyte antigen (HLA) loci (40). In a study, 4 out of 11 patients of low-risk cervical intraepithelial neoplasia 1 (CIN-1) and 6 among 27 of high-risk CIN-3 patients, were found to have mutated E2 (35). Malignancies associated with HPV infection are the most studied compared to the malignancies caused by other viruses. Since the discovery of HPV as a causative agent of cervical cancer in 1907, preventive measurements have been taken place worldwide. The routine Papanicolaou test (PAP smear) provides extensive information about HPV infection and cervical cancer development. Recently, a vaccine against high-risk types, that is HPV 16 and 18, has been formulated. It is estimated that the development of this vaccine will reduce the annual HPV-associated cancer from 500,000 to 450,000. Moreover, the vaccine will also be helpful to limit the onward HPV transmission (9,73).
Epstein–Barr virus
EBV, also known as human herpes virus-4 (HHV-4), is a large double-stranded DNA virus, which was discovered to be the first oncogenic virus. Cells involved in EBV infections are B lymphocytes and epithelial cells (1). Usually EBV infection occurs in childhood, being asymptomatic. In some adult cases, the infection leads to lymphomas like non-Hodgkin's lymphoma and Burkitt's lymphoma (45). EBV infection may cause either lytic or latent infection. Lytic infection involves both B cells and epithelial cells, while latent infections are mostly limited to B cells. EBV expresses its entire genome in lytic infections, producing a high titer of viral oncogenes, which further kill the host cells. In latent infections, EBV partially expresses its genome instead of the whole-genome expression and causes host cell transformations (72).
Latent membrane protein 1 (LMP-1) acts as an EBV oncogene mimicking active CD40 receptor, a tumor necrosis factor receptor (TNFR), resulting in uncontrolled cellular proliferations (33,43). Gastric carcinomas by EBV include almost 10% of the whole gastric carcinoma patients. In a study of 465 gastroesophageal carcinoma patients, 14 cases were found to be EBV positive. They were also tested for EBV-encoded small RNAs (EBER) by in situ hybridization techniques. EBV causes DNA hypermethylation and Janus kinase 2 (JAK2) amplifications (23). Examination of gastric carcinoma with lymphoid stroma (GCLS) patients was done for concluding its association with EBV, and more than 85% patients were found to be EBV positive (37). Similarly, EBV miRNA profiling in gastric carcinoma patients was also performed and dominating role of EBV miRNA-BART4-5p (BamHI A rightward transcripts) in carcinogenesis was observed (57).
EBV infection leads to a latent state in most of the patients and release some proteins like EBV nuclear antigen 1 (EBNA-1), LMP-1, and EBER. EBNA-1 is the only protein that is expressed in all types of EBV-associated malignancies and helps in the viral episomal maintenance, replication, and transformation. Increasing evidences have suggested that EBNA-1 may alter the cellular environment, induce genomic instability, and have the potential to act as an oncogene (25,51). EBNA-2 is another protein released for expressing cellular genes like viral receptors (66). It deregulates c-myc expression and causes increased cellular proliferation. To enter the lytic state, EBV initially expresses all its latent state III proteins (EBNA 1, 2, 3A, 3B, 3C, LP, and LMP1, LMP2 proteins) and blasts the activated B cell to enter the tonsillar germinal center where it downregulates the expression of EBNA proteins 2, 3A, 3B, 3C, and LP as shown in Figure 3. The continued expression of latency state II proteins (EBNA1, LMP1, and LMP 2) in the germinal center develops the B cell to memory B cell, which exits in the germinal center to the circulating blood, where it expresses no viral protein except EBNA 1 (Latency I) protein during cell mitosis. EBNA 1 exploits host cell machinery to ensure the duplication of EBV in each daughter cell. When latently infected B cells return tonsil, they terminally differentiate into plasma cell to initiate lytic cycle (51).
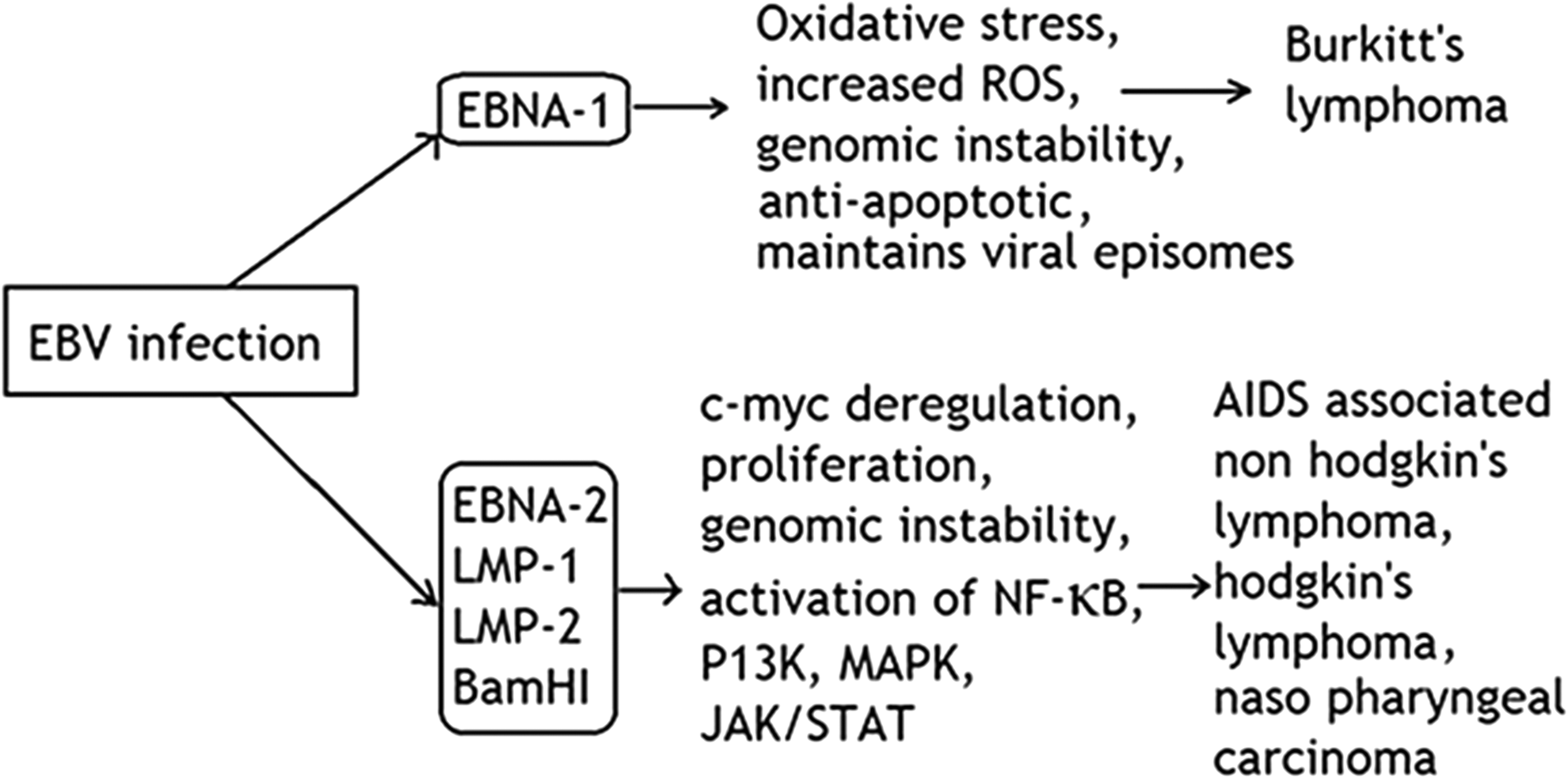
EBV induced oncogenic mechanism: EBV infection causes the expression of EBNA1 gene that inudes host cell genome instability and exploits host cell mechinary for its own genome replication. The other proteins like EBNA2, LMP1, LMP2, and BamH1 are expressed at different stages of EBV latency and alters multiple signaling pathways. EBV, Epstein–Barr virus.
Hepatitis B virus
HBV is a partially double-stranded DNA virus. HBV was diagnosed as a serum-borne infective jaundice in 1940 (11). HBV is highly contagious and may be transmitted by exposure to HBV-infected blood and other body fluids through breaks in cutaneous and mucosal membrane. HBV infection is usually asymptomatic in adults, while it becomes chronic in young children or neonates (53). Chronicity with HBV has a major role in the development of hepatocellular carcinoma (HCC), and antiviral drugs are used to reduce HBV DNA levels to mitigate the possibility of HCC. HBV integrates its DNA into the host cellular genome causing mutations and accelerating liver inflammation (38). The precise pathologic mechanism of HBV infection is not completely understood. None of HBV proteins has been directly linked with acute oncogenic activity as the process of HCC development is a multistep complicated process that spans several decades (26). HBV encoded X antigen (HBx), a transcriptional activator of host cellular genes that is involved in HCC development. HBx activates specific cyclins, cyclin-dependent pathways, JAK/STAT pathway, and mitogen-activated protein kinase (MAPK) pathway leading to liver carcinogenesis due to interruption of tumor suppressor genes as shown in Figure 4 (17).
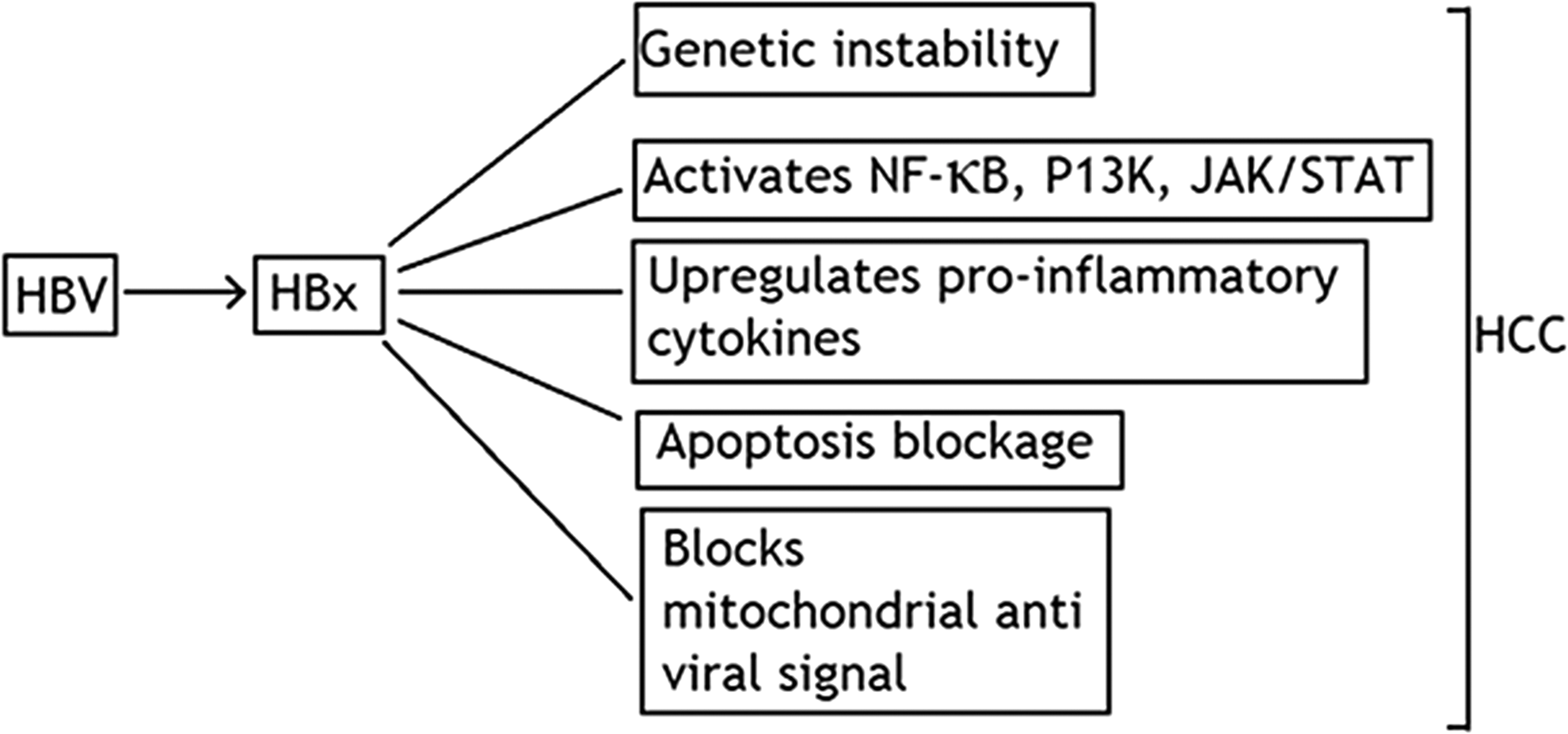
Role of HBx in HCC development: Hepatits B X-antigen induces host cell genome instability and altered expression of various cytokines leading to antiviral state. HCC, hepatocellular carcinoma.
A recent report of mice studies has shown the oncogenic role of HBx protein in the induction of HCC. Only HBx does not cause the cancers, but c-myc involvement leads to the development of cancer. HBx protein only acts as a promoter in tumor induction (60).
HBx causes p53 suppression that promotes the inactivation of pRb and also downregulates various CDK inhibitors (70). The involvement of tumor-associated fetal protein (AFP) action in HBx generating oncogenesis is also reported. The study on 614 HCC patients revealed that AFP is an intracellular signaling molecule acting as a probable participator in HBx inducing carcinogenesis (71). It is reviewed that recurrence of tumors is a dominating obstacle for diagnostic improvement of HBV-related HCC. Major viral factors for hepatic carcinoma recurrence include HBsAg, HBeAg, HBV DNA levels, and HBcAg (48).
Kaposi's sarcoma-associated Herpes virus
KSHV is a double-stranded DNA virus. Simliar to EBV, KSHV also causes either latent or lytic infections. It may infects a range of the cells including B cells, endothelial cells, and monocytes. KSHV leads to three disorders in humans namely primary effusion lymphoma (PEL), Kaposi's carcinoma, and Multicentric Castleman's disease (MCD) (59). Transmission route of KSHV is yet unknown. It either transmits sexually or through parenteral transmission pathways. KSHV infections are usually asymptomatic, but coinfection with HIV leads to Kaposi's sarcoma (21). LANA plays a key role in KSHV-associated oncogenesis. It causes infected cell proliferations by inactivating tumor suppressors, p53 and pRb. LANA is multifunctional as it interacts with various host proteins, including p53, pRb, and glycogen synthase kinase 3β (GSK-3β) as shown in Figure 5. Interaction with GSK-3β increases the level of beta-catenin that leads to tumor progression. Inactivation of p53 and pRb blocks cell cycle arrest and causes uncontrolled proliferations (20). LANA also activates the expression of human telomerase reverse transcriptase (hTERT) leading to limitless proliferations (65).

Role of LANA in KSHV-induced oncogenesis: KSHV-associated LANA inactivates p53 and pRb to take control over tumor suppression and cell cycle, respectively. LANA also binds with GSK-3β to initiate tumor angiogenesis. KSHV, Kaposi's sarcoma herpes virus.
RNA Oncoviruses and Their Targets
Among RNA viruses, HCV and HTLV-1 are associated with human malignancies. Both viruses adopt different oncogenic mechanisms. In some cases, overproduction of oncogenic materials stimulates cellular proliferations (54), while in other cases viruses integrate their genomes near cellular growth stimulating genes to initiate cellular transformations. Some RNA viruses have Tax protein that activates the expression of cellular genes (14). Tax modulations promote malignant transformations through host cell cycle disruptions and uncontrollable divisions. HTLV basic leucine zipper protein (HBZ) promotes cell proliferations and suppresses Tax-mediated transactivation. Major histocompatibility class-1 (MHC-1), Signal Transducer and Activator of Transcription-5 (STAT-5), and hTERT are the host cellular targets for RNA viruses (5).
Hepatitis C virus
HCV is a negative single-stranded RNA virus that was discovered in 1989 by Houghton and colleagues. There are more than 200 million HCV-infected patients worldwide. Twenty-five percent of chronically HCV-infected patients develop cirrhosis and other liver complications. Approximately, 20% patients enter the stage of HCC (61). HCV is also considered to possess a dominating role in HCC generation. HCC is the fifth most common cancer worldwide and thought to be the third leading cause of death (15). HCV infection may be transmitted through blood transfusions, blood products, organ transplantations, and unsterilized injections.
HCV infection stimulates continuous growth and upregulates telomerase expression resulting in immortalization (49). HCV interferes with the promyelocytic leukemia (PML) tumor suppressor protein, which is the cause of chronicity (27). HCV also upregulates DNA methyl transferases, which further block tumor suppressor genes leading to HCC (62). HCC and HCV association was first reported in 1990 (34). HCV core proteins generate chronic oxidative stress causing chromosomal and mitochondrial DNA instability (29). Core protein activates cellular oncoproteins and NF-κB cell signaling pathways. It also causes p53 & pRb inactivation to initiate genomic instability and uncontrollable cellular proliferations (58). HCV nonstructural proteins NS5A and NS3 directly induce HCC by altering host expression and promote liver cell proliferation (31). NS5A causes suppression of immune responses, inactivation of tumor suppressors, loss of apoptosis, and disrupted homeostasis as shown in Figure 6.

HCV induced hepatocellular carcinoma: HCV core protein, NS2, NS3, NS5A, and NS5B are majorly involved in progression of HCV-induced HCC.
Human T Lymphotropic Virus type 1
The discovery of adult T cell leukemia in Japan by Takatsuki and colleagues in 1979 set the stage for the discovery of first human retro-oncovirus (64). In 1980, a series of experiments were performed by the scientific community to isolate human oncoretrovirus, but finally, Robert Gallo was successful in the identification of a novel human retrovirus from cultured human T cell lymphoma cells and were termed as human T cell lymphotropic cells (HTLV-1) (47). There are approximately 10–20 million HTLV-1-infected patients and its precise mechanism of pathogenesis is still a challenge for researchers.
HTLV-1 is a single-stranded RNA virus harboring several genes like gag, pol, env, tax, and two long terminal repeats (72). There is little information about how HTLV-1 causes cancer, but it is cleared that HTLV-1 Tax protein plays a central role in carcinogensis rather than insertional mutation that is a typical feature of animal retroviruses. HTLV-1 Tax protein and HBz play an essential role by interfering with several regulator pathways, cell cycle, and cellular transformation (69). HTLV-1 is associated with adult T cell leukemias and neurodegenerative disorders. Tax protein mainly targets cytotoxic T cells inducing genome instability and stimulating cell proliferations. Tax protein indirectly interacts with host genome and causes modulation of transcriptional factors and activates CDK and hTERT (3). HBz directly binds to the host genome and activates cancer hallmarks and hTERT expression as shown in Figure 7 (52).

HTLV-1 Tax and HBz gene induced oncogenesis: Tax protein alters several signaling pathways and takes control over cell cycle. HBz causes continuous cellular proliferation and hTERT expression. hTERT, human telomerase reverse transcriptase.
Immortal CD4+ and CD8+ cells are found in HTLV-1 chronic infections. The infection is endemic in Africa, Japan, and Central and South America (8). Till date, there is no effective vaccine available against HTLV-1 infection and the treatment is restricted only to the management of opportunistic infections that may be the result of immune suppression by HTLV-1 (63).
Conclusion
Oncogenic mechanisms of some major viruses have been well described. Oncoviruses act in different ways depending upon the factors of host. Some small genomic viruses act by integrating in the host cell genome and causing mutations in it. Some retroviruses activate the expressions of proto-oncogenes inducing several different malignancies. Oncoviruses activate already existing switched-off genes inside the body and boost their expressions, which leads to cell cancerous state and uncontrolled proliferations.
The worldwide history shows that among all human cancers, approximately 20% cancers are caused by oncoviruses (73). Viruses are considered a primary source of cancer growth and must not be ignored; otherwise, advancement in proper treatment against cancers would be no more possible. There are viruses that are addressed to be cancer causing for humans, but still there is no sufficient data to create a consensus regarding their etiological role in cancer development. Further studies on the viral oncogenic mechanisms on the basis of their cellular and molecular roles may be helpful in designing effective cancer therapeutics. There is a need to construct preventive actions, vaccines, and drugs against viruses. Vaccines for HPV and HBV have got milestones in the field of medicine and inspire many researchers to work on such protecting and lifesaving vaccines against other oncoviruses. HPV vaccines named Cervarix and Gardasil are reported to be, approximately, 100% effective against HPV 16 and HPV 18. Production of different therapeutics that venture virus-specific proteins, for example, the development of immunotherapies, targeted monoclonal antibody, and so on, must be an important future goal.
Footnotes
Acknowledgments
The study is supported by Faculty of Health and Allied Sciences, Imperial College of Business Studies, Lahore, Pakistan.
Author Disclosure Statement
All the authors declare that there are no conflicts of interest in publication of this review article.