
Abstract
Vaccination is a proven intervention against human viral diseases; however, success against Herpes Simplex Virus 2 (HSV-2) remains elusive. Most HSV-2 vaccines tested in humans to date contained just one or two immunogens, such as the virion attachment receptor glycoprotein D (gD) and/or the envelope fusion protein, glycoprotein B (gB). At least three factors may have contributed to the failures of subunit-based HSV-2 vaccines. First, immune responses directed against one or two viral antigens may lack sufficient antigenic breadth for efficacy. Second, the antibody responses elicited by these vaccines may have lacked necessary Fc-mediated effector functions. Third, these subunit vaccines may not have generated necessary protective cellular immune responses. We hypothesized that a polyvalent combination of HSV-2 antigens expressed from a DNA vaccine with an adjuvant that polarizes immune responses toward a T helper 1 (Th1) phenotype would compose a more effective vaccine. We demonstrate that delivery of DNA expressing full-length HSV-2 glycoprotein immunogens by electroporation with the adjuvant interleukin 12 (IL-12) generates substantially greater protection against a high-dose HSV-2 vaginal challenge than a recombinant gD subunit vaccine adjuvanted with alum and monophosphoryl lipid A (MPL). Our results further show that DNA vaccines targeting optimal combinations of surface glycoproteins provide better protection than gD alone and provide similar survival benefits and disease symptom reductions compared with a potent live attenuated HSV-2 0ΔNLS vaccine, but that mice vaccinated with HSV-2 0ΔNLS clear the virus much faster. Together, our data indicate that adjuvanted multivalent DNA vaccines hold promise for an effective HSV-2 vaccine, but that further improvements may be required.
Introduction
B
Antibodies directed against virion glycoproteins are an important mechanism by which the immune system combats viral infections. These antibodies can prevent entry of the virus into cells by conventional neutralization (i.e., physically blocking the interaction of the virus glycoprotein with its cellular receptor), and they can eliminate viruses and/or infected cells through Fc-mediated effector mechanisms (15,26,41,73). Unfortunately, protein subunit vaccines targeting the HSV-2 entry receptor glycoprotein D (gD) have thus far failed in human trials (14,27,84 –86). At least three factors may have contributed to the failures of gD-based protein subunit vaccines. First, immune responses directed against gD may lack sufficient antigenic breadth (42). Second, the antibodies elicited by these subunit vaccines may have lacked necessary Fc-mediated effector functions such as antibody-dependent cellular cytotoxicity (ADCC) and complement fixation. Third, gD may not generate protective cellular immune responses that could be necessary for combating HSV-2.
In addition to gD, six other viral glycoproteins are primarily involved in HSV-2's cellular entry and cell-to-cell spread (3,28,35). HSV-2 entry into cells occurs through a series of steps that are mediated by a multi-component glycoprotein structure on the surface of HSV-2 virions that includes glycoprotein B (gB), gC, gD, and the gH/gL heterodimer (3,21,28,35,37). gD serves as the main attachment protein for HSV-2, whereas gB and gC assist in tethering virions to target cells (3,28,35). Fusion is mediated by gB, gD, and the gH/gL heterodimer (3,28,35), which are essential for entry (3,6,21,28,35). gC increases the efficiency of fusion, but it is not strictly required for HSV-2 entry (3,28,35). Cell-to-cell spread requires gD and the gE/gI heterodimer (3,28,35). Of these glycoproteins, gD and gB have been well studied for their potential to serve as HSV-2 vaccine antigens (23,27,29,55), whereas the remaining HSV-2 glycoproteins have received less attention (23,27,29,55).
Although surface glycoproteins have garnered the most attention as vaccine immunogens, intracellular HSV-2 proteins that are designed to elicit protective cellular immune responses have also been tested with some success (18,45,60). Surprisingly, the immunodominant antigens (based on antibody recognition) that were identified after vaccination with a highly effective attenuated HSV-2 viral vaccine denoted HSV-2 0ΔNLS (43,44) were not viral coat antigens, but were intracellular viral antigens (40). The most humoral immunodominant HSV-2 0ΔNLS antigens were found to be VP5, ICP8, and ICP10 (40). These results indicate that such intracellular viral proteins may be important antigens and, therefore, worth evaluating as vaccine immunogens.
Of the many vaccination strategies developed to date, DNA vaccines are particularly exciting, because they provide a unique platform that can be tailored to elicit high-quality anti-viral antibodies and cellular immune responses and they do not suffer from anti-vector immunity. The first generation of DNA vaccines suffered from two major drawbacks that reduced their efficacy in humans. First, DNA vaccines delivered intramuscularly (i.m.) were not efficiently expressed by host cells and, in turn, elicited weak immune responses (2,10,12,46,57,58,71,76,77,89). This issue has been resolved with highly efficient DNA vaccine delivery strategies (2,10,12,46,57,58,71,76,77,89). For example, in vivo electroporation (EP) of plasmid DNA increases antigen expression in vaccinated tissues >100-fold (2,10,12,46,57,58,71,76,77,89), and it has yielded second-generation DNA vaccines that have been encouraging in non-human primates and in human trials (10,12,36,46,57).
Second, DNA vaccines do not adequately stimulate innate immune responses that are required to trigger potent, long-lived adaptive immune responses to foreign antigens. Similar to protein subunit vaccines, DNA vaccines do not contain endogenous “danger” signals that are capable of activating the innate immune system. It was originally believed that plasmid DNA would be a TLR9 agonist and would provide innate immune activation on vaccination. However, it was later found that plasmid DNA is not an effective TLR9 agonist (11). EP evokes an inflammatory response; however, it is weak compared with that caused by natural infections, and it dissipates long before antigen expression wanes (24). Thus, the first and second generations of plasmid DNA vaccines effectively lack an adjuvant. This issue is being resolved in third-generation vaccines by including adjuvant plasmids with vaccine plasmids (1,4,13,31, 48,49,54,56,72,78,91,92).
We and others have used the DNA adjuvant interleukin 12 (IL-12) to enhance the immunogenicity, and to skew the evoked immune responses toward a T helper 1 (Th1) phenotype (25,33,34,58,75,91). Clinical study HVTN-080 demonstrated that the inclusion of IL-12 can double the CD4+ and CD8+ T cell response rate in vaccines over EP alone to >80% (52). We also found that the administration of IL-12 induces polyfunctional T cells that are known to correlate with protection from viral infections (25,33,58,75).
In the current study, we tested several hypotheses. Hypothesis 1: Delivering HSV-2 gD as a DNA vaccine adjuvanted with IL-12 by EP will evoke superior protection from HSV-2 challenge compared with the same immunogen delivered as a recombinant protein mixed with adjuvants. Hypothesis 2: Inclusion of additional glycoproteins with gD in a DNA vaccine will enhance protection over that offered by gD alone. Hypothesis 3: The intracellular antigens VP5, ICP8, and ICP10 either by themselves or combined with gD will elicit protection from challenge. Hypotheses 1 and 2 were supported by our studies, whereas the third hypothesis was refuted. In summary, our data demonstrate that mice immunized with plasmid DNAs expressing IL-12 and several HSV-2 glycoproteins, but not intracellular proteins, provided similar protection to that afforded by the live attenuated HSV-2 vaccine HSV-2 0ΔNLS (43,44) in terms of enhanced survival, reduced weight loss, and/or increased weight regain and elimination of visible clinical symptoms. However, HSV-2 0ΔNLS cleared the viral infections much faster than the DNA vaccines. We, therefore, conclude that the DNA approach to HSV-2 vaccine development is promising but still has room for improvement. Possible improvements are discussed.
Results
A gD-based DNA vaccine is more protective than a gD-based protein vaccine in a high-dose challenge mouse model
In our first study, we sought to determine 1–whether a DNA vaccine expressing the full-length HSV-2 gD and the adjuvant IL-12 would elicit protection in immunized mice that was greater than that afforded by a vaccine consisting of a truncated recombinant gD protein adjuvanted with alum and monophosphoryl lipid A (MPL) (43) and 2–whether the HSV-2 0ΔNLS humoral immunodominant intracellular viral antigens VP5, ICP8, and/or ICP10 (40) would elicit protection as DNA immunogens in a high-dose challenge model. As a “benchmark” for robust protection, a live-attenuated HSV-2 0ΔNLS vaccine was included for comparison (43,44). Mice immunized with empty plasmid DNA served as a mock-immunized, naive control group. Each group of mice was immunized on days 0 and 21. The DNA vaccines were delivered by using a low-dose DNA prime (delivered i.m. without EP)/high-dose DNA boost (delivered i.m. with EP) regimen that was shown to improve immune responses (20). After allowing 2 months for immune responses to return to a resting state, all mice were treated with medoxyprogesterone on days 74 and 78 (43,53); then, they were vaginally challenged on day 81 with 500,000 pfu HSV-2 strain MS (∼2,000 times the lethal dose [LD50]).
On days 1 and 3 post-challenge, vaginal HSV-2 shedding was measured in all mice. Mock-immunized mice shed the highest titers of HSV-2 after vaginal challenge, which were an average 16,200 and 69,500 pfu on days 1 and 3 post-challenge, respectively (Fig. 1A). HSV-2 0ΔNLS-immunized mice shed an average 157-fold and >1,000-fold less HSV-2 on days 1 and 3 post-challenge (Fig. 1A) (p < 0.001 on both days). Strikingly, only one of the nine HSV-2 0ΔNLS-immunized mice shed detectable HSV-2 on day 3. Mice immunized with the gD+alum/MPL subunit vaccine shed an average 9.1 and 2.7-fold less HSV-2 days 1 and 3, respectively (Fig. 1A), but these reductions did not reach significance (Fig. 1B). The results from the HSV-2 0ΔNLS and gD+alum/MPL subunit groups are consistent with those of previously published studies (43). Mice immunized with the IL-12 adjuvanted gD DNA vaccine shed an average 24- and 7-fold less HSV-2 on days 1 and 3, respectively (Fig. 1A), and these reductions were significant (Fig. 1B). These mice also shed significantly less HSV-2 on day 3 compared with the mice immunized with the gD subunit vaccine (Fig. 1A, B).

Virus shedding post-challenge in study 1. Groups of 10 mice were vaccinated on days 0 and 21 and were treated with medoxyprogesterone 7 and 3 days before vaginal HSV-2 challenge on day 81 with 500,000 pfu of HSV-2 MS. HSV-2 shedding from vaginas was determined on days 1 and 3 post-challenge.
Mice immunized with VP5, ICP8, or ICP10 had reduced levels of viral shedding on days 1 and 3 post-challenge; however, the only significant reduction in shedding was from mice immunized with VP5 on day 1 post-challenge (Fig. 1B). Mice immunized with a combination of DNAs expressing gD, VP5, ICP8, and ICP10 had significantly reduced shedding on days 1 and 3 post-challenge compared with the mock-immunized group (Fig. 1A, B) and reduced shedding compared with the gD DNA-alone immunized group (Fig. 1B); however, the reduction in shedding compared with the gD DNA-alone immunized group was only significant on day 1 post-challenge (Fig. 1B).
Together, these results indicate that the gD DNA vaccine is significantly better than the gD subunit vaccine for reducing viral shedding, but that the HSV-2 0ΔNLS vaccine is dramatically better for reducing viral shedding than the gD DNA vaccine. These results also indicate that the intracellular antigens VP5, ICP8, and ICP10 are poor immunogens for reducing shedding by themselves but may slightly enhance the effectiveness of the gD DNA vaccine. Of note, the dose of gD delivered to the gD+VP5+ICP8+ICP10 group was only one-fourth dose delivered to the gD DNA-alone group.
Between days 0 and 14 post-challenge, mice were monitored for survival. Any animal losing more than 25% of its starting body weight was humanely euthanized and considered to have succumbed to their infections. All of the mock-immunized animals succumbed to their infections by day 10 post-challenge (Fig. 2A). All of these animals also displayed visible symptoms of infection before death, such as lethargy, hunched back, and piloerection (data not shown). Consistent with previous studies using this virus challenge dose (43), 80% of the mice immunized with the gD+alum/MPL subunit vaccine succumbed to their infections by day 13 post-challenge (Fig. 2A), and this increase in survival over the mock-immunized group was significant (Fig. 2B). All of the animals in this group, including the two survivors, also displayed all of the same visible symptoms of infection as the mock-immunized animals (not shown). By stark contrast, all of the mice immunized with the HSV-2 0ΔNLS and gD DNA vaccines survived the challenge (Fig. 2A) and none of the mice in either of these groups displayed visible signs of infection (not shown). The increases in survival in these two groups were highly significant compared with both the mock-immunized group and the gD subunit-vaccinated group (Fig. 2B).
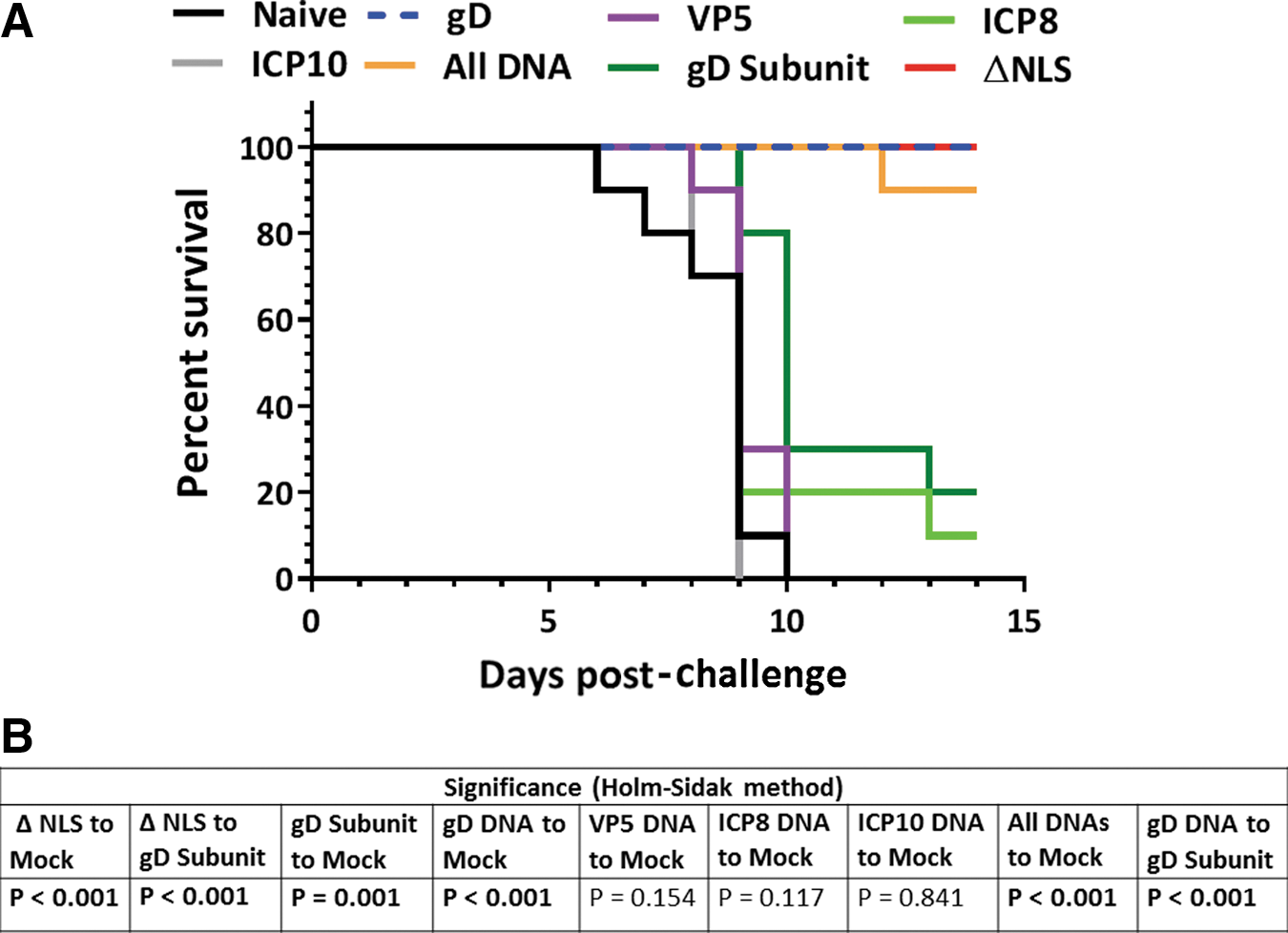
Survival post-challenge in study 1. Groups of 10 mice were vaccinated on days 0 and 21 and were treated with medoxyprogesterone 7 and 3 days before vaginal HSV-2 challenge on day 81 with 500,000 pfu of HSV-2 MS. Mice were monitored for survival and weight loss for 14 days post-challenge. Mice losing ≥25% of their pre-challenge body weights were humanely euthanized and were considered to have succumbed to their infections.
All of the mice immunized with the VP5 or ICP10 DNA vaccines died after challenge, and only one mouse in the ICP8 DNA group survived (Fig. 2A). All of the mice in these groups, including the sole survivor in the ICP8 group, also displayed visual symptoms of infection similar to the mock-immunized mice (not shown). Ninety percent of the mice in the gD+VP5+ICP8+ICP10 (all DNA group) group survived the challenge (Fig. 2A); however, half of the mice in this group displayed visual symptoms of disease, although in three of these five mice the symptoms were less severe than those of mock-immunized mice (not shown).
Together, these results indicate that both the gD DNA vaccine and the HSV-2 0ΔNLS vaccine are dramatically better for increasing survival and reducing visible symptoms than the gD subunit vaccine. The results also indicate that the VP5, ICP8, and ICP10 antigens did not significantly improve survival and that addition of these antigens to the gD DNA vaccine actually reduced the effectiveness of gD by decreasing survival (although only by 10%) and by worsening clinical symptoms.
Between days 0 and 14 post-challenge, the weight of each mouse was also measured. Mock-immunized mice rapidly lost weight after HSV-2 vaginal challenge until they succumbed to their infections or were sacrificed due to the loss of >25% of their initial body weight (Fig. 3). By contrast, most HSV-2 0ΔNLS-immunized mice maintained weight or gained weight after HSV-2 vaginal challenge (Fig. 3A). Eight of the 10 mice immunized with the gD+alum/MPL subunit vaccine rapidly lost weight after HSV-2 vaginal challenge until their death or sacrifice, whereas the 2 surviving mice lost weight for 10 days and then recovered (Fig. 3A). Of note, the reduction in weight loss of the gD subunit-immunized mice compared with the mock-immunized mice on day 8 post-challenge (the last day when 70% of the mock-immunized mice remained alive) was significant (Fig. 3C). Interestingly, the gD DNA-immunized mice exhibited considerably less weight loss between days 0 and 14 post-challenge than the gD subunit-immunized mice (Fig. 3A), and the differences in the weight loss between these groups on days 8 and 9 post-challenge (the last days when 80% of the gD subunit-immunized mice remained alive) were highly significant (Fig. 3C).

Weight loss and regain post-challenge in study 1. Groups of 10 mice were vaccinated on days 0 and 21 and were treated with medoxyprogesterone 7 and 3 days before vaginal HSV-2 challenge on day 81 with 500,000 pfu of HSV-2 MS.
Mice immunized with VP5 or ICP10 DNAs had weight loss profiles that were not improved compared with the mock-immunized controls (Fig. 3B), and the mice in the ICP10 group tended to lose weight even more quickly than the controls since their weight loss was significantly more profound than the controls on day 6 post-challenge (p = 0.018). By contrast, the animals immunized with ICP8 tended to lose less weight early after infection, since they had lost significantly less weight than mock-immunized controls on days 6 and 7 post-challenge (Fig. 3B) (p = 0.034 and p = 0.019, respectively). The sharp rebound in weight regain in this group was the result of two mice surviving until day 12, one of which (the sole survivor) rapidly regained weight.
Together, the results of this first study indicate that the IL-12-adjuvanted gD DNA vaccine is significantly better than the gD subunit vaccine for reducing virus shedding, improving survival, and reducing weight loss post-infection, but that the HSV-2 0ΔNLS vaccine is superior to the gD DNA vaccine, especially for reducing virus shedding. The results also indicate that of the intracellular DNA antigens, only ICP8 provided a small amount of protection whereas ICP10 actually worsened disease. Finally, the results indicate that combining the intracellular antigens with gD DNA slightly improves early virus shedding but worsens clinical symptoms.
A gD-expressing DNA vaccine elicits an IgG2A-skewed antibody response with higher neutralization capacity that correlates with improved protection against HSV-2 vaginal challenge
The gD DNA vaccine and the gD subunit vaccine are based on similar gD antigens (i.e., full-length gD vs. truncated gD, respectively), but they elicit substantially different levels of protection against HSV-2 vaginal challenge. We, therefore, determined whether these two vaccine regimens elicited quantitative and/or qualitative differences in the antibody responses to gD. To this end, serum was obtained from individual mice in each of the immunization groups on day 77 before challenge, and the titers of total IgG and the IgG1, and IgG2a isotypes directed against gD were determined. The sera of mock-immunized mice showed negligible binding of total IgG, IgG1, and IgG2A to gD, and they defined the background of the antibody-capture enzyme-linked immunosorbent assay (ELISA) (Fig. 4A). Mice immunized with the gD in alum/MPL subunit vaccine had the highest titer of total IgG against gD (Fig. 4A), but the differences in anti-gD total IgG between this group and the DNA and HSV-2 0ΔNLS groups were not significant (Fig. 4C). Stark differences were, however, observed in the isotypes of the gD-specific antibodies evoked by the different vaccine regimens. This is relevant, because IgG1 is considered a Th2-associated isotype, whereas IgG2a is considered a Th1-associated isotype with a broader range of effector functions, including ADCC and complement fixation (50,59,62).
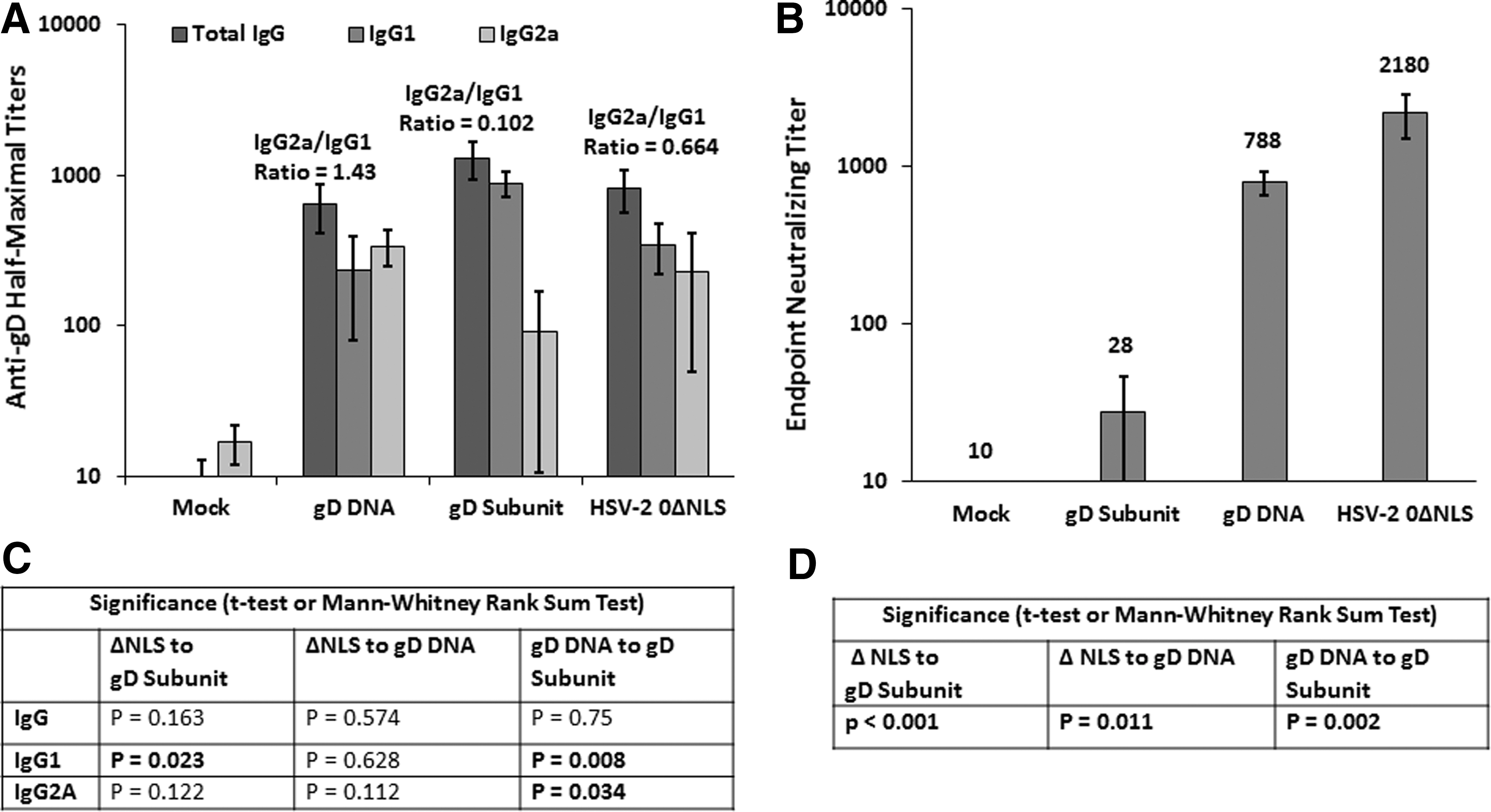
Anti-gD antibody magnitude, isotype distribution, and neutralization capacity before challenge in study 1. Mice were vaccinated on days 0 and 21, and individual serum samples were obtained on day 77 (4 days before challenge). The half-maximal titers of gD-specific total IgG and the IgG1 and IgG2A isotypes were determined on individual serum samples by ELISA.
The antibody responses of mice immunized with gD in alum/MPL were dominated by the IgG1 isotype with an average IgG2a/IgG1 ratio of only 0.1 (Fig. 4A). In fact, only one mouse immunized with the gD subunit vaccine had an IgG2a titer above background (IgG2A titer 734 and IgG1 titer 967). Interestingly, this mouse (with an IgG2a/IgG1 ratio of 0.75) was: 1–one of the two survivors in that group, 2–lost the least weight in that group, and 3–shed the least HSV-2 on day 3 post-challenge relative to the other nine mice in this immunization group (not shown). In contrast to this group, the gD DNA evoked antibodies that were skewed toward the IgG2a isotype with an average IgG2a/IgG1 ratio of 1.4 (Fig. 4A), and the mean IgG2A titer of the gD DNA group was significantly greater than that of the gD subunit group (Fig. 4C). The mean IgG1 titer of the gD DNA group was also significantly lower than that of the gD subunit group (Fig. 4C). The HSV-2 0ΔNLS vaccine evoked modestly more IgG1 than IgG2a, with an average IgG2a/IgG1 ratio of 0.66 (Fig. 4A). Together, these results indicate that there was little difference in the magnitudes of the total anti-gD IgG antibody responses evoked by these different vaccines, but that the gD DNA vaccine evoked a much higher IgG2a/IgG1 ratio than the gD subunit vaccine.
Serum taken before challenge was also tested for neutralization of HSV-2. The HSV-2 0ΔNLS-immunized animals had the highest neutralizing titers with a mean endpoint titer of 2,180 (Fig. 4B) that was significantly higher than the mean titers of the gD subunit and gD DNA-vaccinated groups (Fig. 4D). The gD DNA-vaccinated animals also had robust neutralizing titers with a mean titer <3-fold lower than that of the HSV-2 0ΔNLS-immunized animals, but 28-fold higher than the mean titer of the gD subunit-immunized mice (Fig. 4B). This difference in neutralization titers between the gD DNA and gD subunit-immunized mice was also highly significant (Fig. 4D). Together, these results indicate that neutralizing titers and IgG2a/IgG1 ratios are associated with protection from challenge. It should be noted that the neutralization assay performed was a complement-dependent assay, meaning that neutralization in this assay is, at least in part, due to complement fixing antibodies.
HSV-2 0ΔNLS antiserum-screening identifies six other viral glycoproteins of vaccine interest
We were interested in determining whether the efficacy of a DNA-based HSV-2 vaccine could be enhanced by the inclusion of plasmid DNAs that expressed at least one other, HSV-2 glycoprotein antigen involved in cell entry and/or cell-to-cell virus spreading. To test this hypothesis, codon and RNA-optimized nucleic acid sequences of gB, gC, gE, gH, gI, and gL were synthesized and cloned into the DNA vaccine plasmid pPBS. To verify that each plasmid construct expressed the intended antigen and to get an idea of the magnitude of targeting by serum from HSV-2 0ΔNLS-immunized mice, human 293 cells were transfected with each glycoprotein-expressing plasmid alone, or were transfected as pairs of plasmids that expressed each subunit of the gE/gI or gH/gL heterodimers (Fig. 5). Twenty-four hours later, cells were stained for cell-surface expression of the glycoprotein antigens with pooled HSV-2 0ΔNLS antiserum and an FITC-labeled anti-mouse IgG secondary antibody. Relative glycoprotein expression levels on live cells were approximated by FITC labeling intensity.

HSV-2 0ΔNLS immune sera recognize seven glycoproteins involved in HSV-2 cell entry and cell-cell spread. Half a million 293 cells plated in individual wells of six-well tissue culture plates were transfected with 1 μg each of the indicated glycoprotein-expressing plasmids or an empty plasmid as a mock control. Twenty-four hours after transfection, cells were incubated with pooled, 293 cell-adsorbed HSV-2 0ΔNLS antiserum, washed, and finally further incubated with an anti-mouse FITC-conjugated secondary antibody. The cells were then washed and run on an FACSCalibur Flow Cytometer. Data were analyzed by using FlowJo software. Black histograms are from mock-transfected cells. Gray histograms are from cells transfected with the indicated glycoprotein plasmids or combination of plasmids.
The rank order of staining intensity was the gH/gL heterodimer > gD > the gE/gI heterodimer > gC > gB (Fig. 5). Cells transfected with the individual gE, gH, or gL-expressing plasmids were not reactive or only weakly reactive, and cells transfected with the gI plasmid were moderately reactive with mouse anti-0ΔNLS antiserum (Fig. 5). By contrast, cells transfected with the gE+gI plasmids or gH+gL plasmids were strongly reactive with the 0ΔNLS antiserum (Fig. 5). This result was expected, since the gE, gI, gH, and gL proteins are not efficiently transported to the cell surface in the absence of their heterodimer partners (3,28). Collectively, these analyses indicate that all of the plasmid DNA constructs expressed their intended HSV-2 glycoproteins, and that six of the seven DNA-expressed viral glycoproteins are moderate to strong antigenic targets of the IgG antibody response generated by the HSV-2 0ΔNLS vaccine.
The addition of gC- or gH/gL-expressing plasmids to a gD-expressing DNA vaccine significantly increases vaccine-induced protection against HSV-2 vaginal shedding
A second mouse study was conducted to test the hypothesis that the efficacy of a gD-based DNA vaccine would be enhanced by the addition of plasmid DNAs expressing other HSV-2 glycoproteins. To this end, mice were immunized with empty plasmid DNA as a mock-immunized control or with HSV-2 0ΔNLS as a positive control for robust protection. The low-dose prime/high-dose boost strategy used in the first study was abandoned in favor of a more traditional high-dose prime/high-dose boost approach (i.e., both the prime and boost immunizations were delivered with EP) to simplify the immunization regimen and in hopes of reducing the efficacy of the gD vaccine to improve our chances of detecting improvements offered by the additional glycoprotein plasmids.
Test groups of mice were immunized on days 0 and 21 with the IL-12 adjuvant plasmid and 14 μg total of pPBS plasmids expressing the following glycoproteins (equal amounts of each glycoprotein-expressing plasmid): 1–gD; 2–gD+gB; 3–gD+gC; 4–gD+gE+gI; 5–gD+gH+gL; or 6–all seven by EP. All mice were then treated with medoxyprogesterone on days 74 and 78, and then were vaginally challenged with 500,000 pfu of HSV-2 strain MS on day 82. Vaginal HSV-2 shedding was again measured on days 1 and 3 post-challenge, and it was also measured on days 5 and 7 post-challenge to better capture the entire course of virus shedding. The mice were monitored for survival, weight loss or weight gain, and visual clinical symptoms for a full 30 days post-challenge rather than 15 days to ensure that the complete disease course was monitored.
Consistent with the first study, mock-immunized mice shed the highest titers of HSV-2 after vaginal challenge, which were an average 5,480, 11,500, 7,600, and 1,464 pfu/vagina on days 1, 3, 5, and 7 post-challenge, respectively (Fig. 6A). Also consistent with the first study, the majority of mice immunized with HSV-2 0ΔNLS (eight of nine) ceased to shed detectable levels of HSV-2 by day 3 post-challenge (Fig. 6A). Relative to mock-immunized animals, mice immunized with gD DNA alone shed an average 3.9-, 8.3-, and 16.7-fold less HSV-2 per animal on days 1, 3, and 5 post-challenge, respectively (Fig. 6A) and the reductions on days 3 and 5 were highly significant (Fig. 6B). All mice immunized with gD DNA alone ceased to shed detectable HSV-2 by day 7 post-challenge (Fig. 6A).
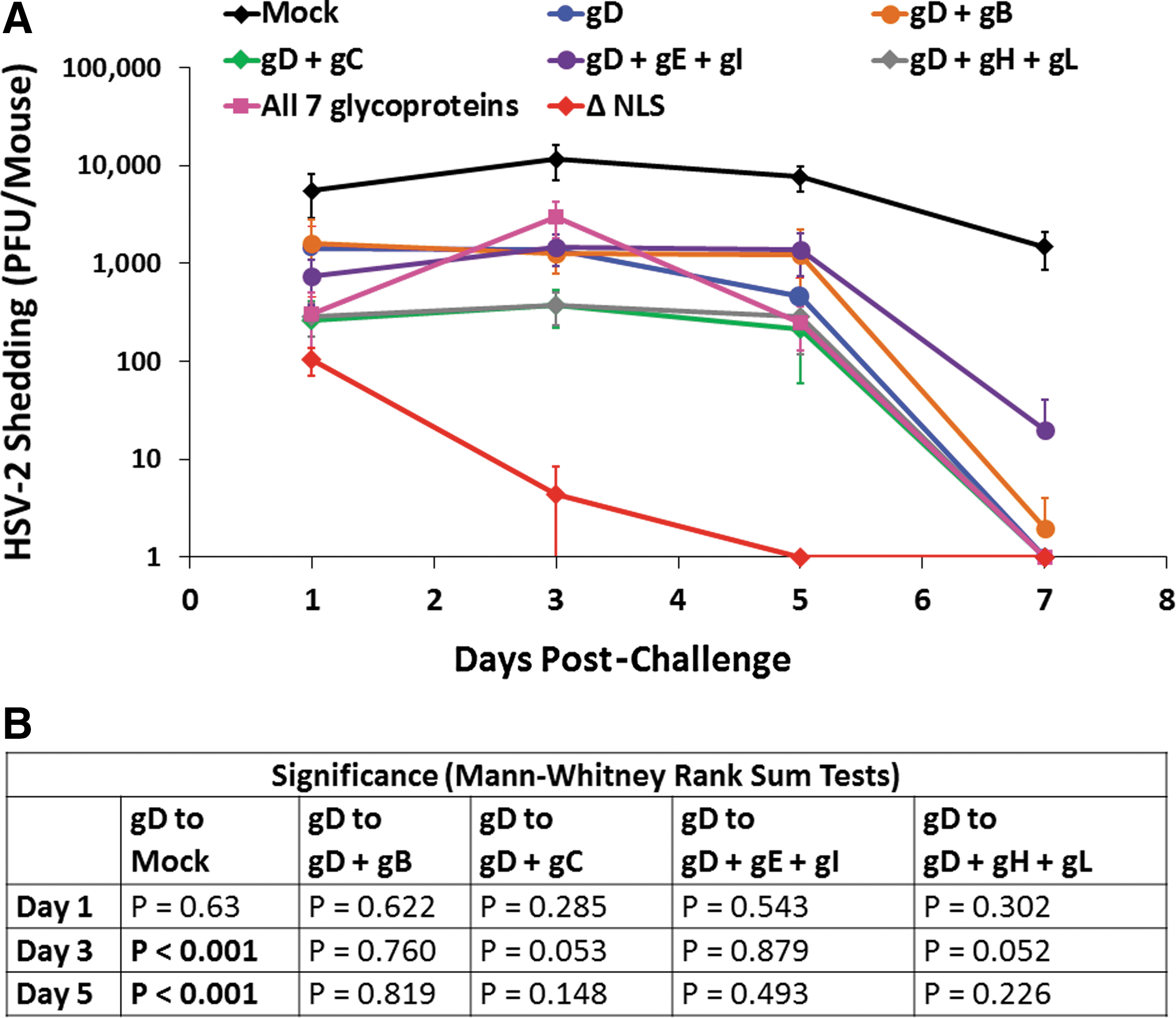
Virus shedding post-challenge in study 2. Groups of 10 mice were vaccinated on days 0 and 21 and were treated with medoxyprogesterone 8 and 4 days before vaginal HSV-2 challenge on day 82 with 500,000 pfu of HSV-2 MS.
The addition of the gB-expressing plasmid, or the gE- and gI-expressing plasmids to the gD plasmid, did not reduce shedding over the gD plasmid alone (Fig. 6A). By contrast, the addition of the gC plasmid, or the gH+gL plasmids to the gD plasmid reduced HSV-2 shedding on days 1, 3, and 5 post-vaginal challenge (Fig. 6A). These reductions were just short of significance on day 3 post-challenge (Fig. 6B). Surprisingly, the group that received all seven glycoprotein-expressing plasmids shed an average 2.2-fold more HSV-2 on day 3 post-challenge than the group that received the gD-expressing plasmid alone (Fig. 6A), although this increase was not significant (Fig. 6B). However, on day 5 post-challenge, these mice shed an average 31-fold less HSV-2 than mock-immunized mice, which was similar to the reduction in HSV-2 vaginal shedding observed in mice immunized with either gD+gC or gD+gH+gL DNAs (Fig. 6A). By day 7 post-challenge, HSV-2 vaginal shedding was only detectable in two of the mice from all of the plasmid DNA-immunized groups combined, whereas all of the mock-immunized mice continued to shed >1,000 pfu on day 7 (Fig. 6A).
Collectively, these data indicate that the addition of gC or gH+gL plasmid DNAs to a gD DNA vaccine enhances protection against HSV-2 in terms of reducing the quantity and/or duration of viral shedding after HSV-2 vaginal challenge but that the reduction in shedding is not nearly as profound as that afforded by the live HSV-2 0ΔNLS vaccine. Specifically, HSV-2 0ΔNLS vaccination caused mice to cease virus shedding 2–4 days quicker than the glycoprotein-expressing DNA vaccines.
The addition of other glycoprotein-expressing DNAs to a gD-based DNA vaccine increases survival and/or protection against weight loss post–HSV-2 challenge
Consistent with the first study, mock-immunized mice rapidly succumbed to their infections or were euthanized after losing ≥25% of their body weight (Fig. 7A). Also consistent with the first study, all of the HSV-2 0ΔNLS-immunized mice survived (Fig. 7A). Eighty percent of mice vaccinated with gD DNA alone survived, whereas 100% of the gD+gB and all seven glycoprotein DNA-immunized mice survived (Fig. 7A). Ninety percent of the gD+gC-immunized mice survived, as did 90% of mice immunized with gD+gH+gL. Seventy percent of the gD+gE+gI-immunized mice survived (Fig. 7A). The survival increases in all DNA-immunized groups were highly significantly improved compared with the mock-immunized (naïve) group; however, the differences in survival between the multiple glycoprotein DNA-immunized groups and the gD DNA alone–immunized group did not reach significance (Fig. 7B).

Survival post-challenge in study 2. Groups of 10 mice were vaccinated on days 0 and 21 and were treated with medoxyprogesterone 8 and 4 days before vaginal HSV-2 challenge on day 82 with 500,000 pfu of HSV-2 MS. Mice were monitored for survival and weight loss for 30 days post-challenge. Mice losing ≥25% of their pre-challenge body weights were humanely euthanized and were considered to have succumbed to their infections.
Mock-immunized mice also rapidly lost weight after challenge until they succumbed to disease, or were euthanized on losing ≥25% of their body weight (Fig. 8A). HSV-2 0ΔNLS-immunized mice gradually lost ∼9% of their body weight until day 12 post-challenge, and they regained weight thereafter (Fig. 8A). The gD DNA alone–immunized mice lost weight more rapidly than the HSV-2 0ΔNLS-immunized mice, reaching a peak weight loss of ∼12% nine days post-challenge, and maintained that peak weight loss through day 15 post-challenge, after which these mice regained body weight (Fig. 8A–D). The gD+gB-immunized mice also lost weight sooner than the HSV-2 0ΔNLS-immunized mice, but they reached the same peak weight loss and regained weight at the same rate as the HSV-2 0ΔNLS-immunized mice (Fig. 8B) and the reduction in weight loss of these mice compared with the gD alone–immunized mice was significant on day 15 post-challenge (Fig. 8E).
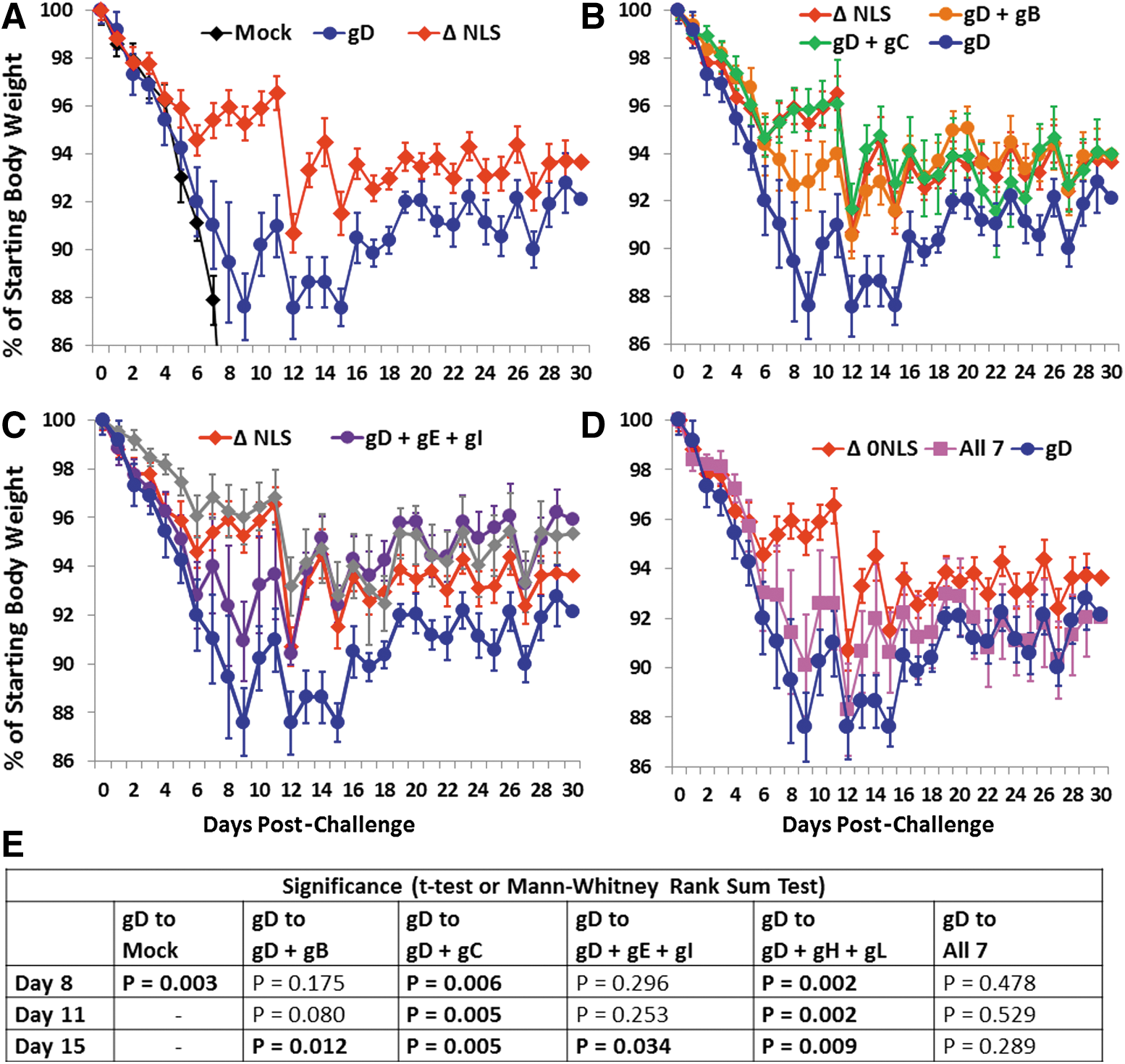
Weight loss and regain post-challenge in study 2. Groups of 10 mice were vaccinated on days 0 and 21 and were treated with medoxyprogesterone 8 and 4 days before vaginal HSV-2 challenge on day 82 with 500,000 pfu of HSV-2 MS.
The gD+gC-immunized mice had weight loss and regain profiles similar to those of mice immunized with HSV-2 0ΔNLS (Fig. 8B), and the reductions in weight loss of these mice compared with those of the gD alone–immunized mice were highly significant on days 8, 11, and 15 post-challenge (Fig. 8E). The gD+gH+gL-immunized mice lost weight less rapidly than the HSV-2 0ΔNLS-immunized mice and regained more weight than the HSV-2 0ΔNLS-immunized mice (Fig. 8C), and the reductions in weight loss of these mice compared with those of the gD alone–immunized mice were also highly significant on days 8, 11, and 15 post-challenge (Fig. 8E). The gD+gE+gI-immunized mice lost weight sooner than the HSV-2 0ΔNLS-immunized mice but reached a similar peak weight loss as the HSV-2 0ΔNLS-immunized mice and regained weight faster than the HSV-2 0ΔNLS-immunized mice (Fig. 8C). The reduction in weight loss of these mice compared with the gD alone–immunized mice was also significant on day 15 post-challenge (Fig. 8E). The mice in the group that received all seven glycoproteins had weight loss and regain profiles that were similar to and not significantly different from those of the gD alone–immunized mice (Fig. 8D, E).
Collectively, these data indicate that the addition of any of the individual glycoproteins or glycoprotein heterodimer pairs to the gD DNA vaccine reduces weight loss after challenge. These data further indicate that addition of the gC plasmid or gH and gL plasmids to the gD vaccine produces improved weight loss/regain profiles that are similar to those provided by the HSV-2 0ΔNLS vaccine.
The magnitudes of the anti-gD humoral and cellular immune responses correlate with the dose of gD DNA delivered
Unfortunately, sera from the second study were not obtained before challenge due to a miscommunication. Therefore, the study was repeated with five mice per group, with the exception that the HSV-2 0ΔNLS group was replaced by a group that received a 2 μg dose of the gD-expressing plasmid (e.g., the dose of gD given in the group that received all seven glycoproteins). The mice from this repeat study were vaccinated in exactly the same way and on the same schedule as the mice from the second study but were sacrificed on day 40 when sera and splenocytes were harvested for anti-gD ELISpots and anti-gD ELISAs.
Figure 9A shows that the magnitudes of the anti-gD antibody responses dropped as the dose of the gD plasmid delivered decreased. For instance, the groups that received gD+gB and gD+gC received half as much gD DNA (7 μg) as the group that received a full dose of gD DNA alone (14 μg), and these groups had half maximal anti-gD titers that were 4.5- to 5-fold lower than the 14 μg gD-alone group. Similarly, the groups that received 2 μg of gD alone or all seven glycoproteins received one-seventh as much gD DNA as the group that received a full 14 μg dose of gD DNA alone, and these groups had half maximal anti-gD titers that were 60- to 93-fold lower than the gD-alone group (Fig. 9A). Importantly, the IgG2a/IgG1 ratios of the anti-gD antibodies were 1.3 or greater for all of the groups that received gD DNA (not shown). This indicates that the skewing of the antibody responses toward the IgG2a isotype is not effected by the dose of the gD plasmid delivered or by the presence of the other glycoprotein antigens.

Anti-gD antibody magnitude, neutralization capacity, and anti-gD IFN-γ ELISpots in a repeat of study 2. Five mice per group were immunized on days 0 and 21 as per study 2 with the exception that the HSV-2 0ΔNLS group was replaced by a group that received a 2 μg dose of the gD-expressing plasmid. All mice were sacrificed on day 40, and sera and splenocytes were harvested for anti-gD ELISAs
Similar to the humoral responses, Figure 9B shows that the magnitudes of the anti-gD ELISpot responses dropped as the dose of gD plasmid delivered decreased. For instance, the groups that received half as much gD DNA (gD+gB and gD+gC) had anti-gD ELISpot responses that were 1.5- to 2.2-fold lower than the gD-alone group, and the groups that received one-seventh as much gD DNA (2 μg of gD alone or all seven glycoproteins) had anti-gD ELISpot responses that were 2.9- to 3.2-fold lower than the gD-alone group. Together, these results indicate that when the total dose of DNA delivered is held constant, the increase in the breadth of the immune responses afforded by including additional glycoprotein immunogens is accompanied by a corresponding decrease in the magnitude of the anti-gD responses.
The sera from this repeat study were also analyzed for HSV-2 neutralization. The serum from the gD in alum/MPL and HSV-2 0ΔNLS groups from the first study cited earlier was also included for comparison. Figure 9C shows that the gD DNA 14 μg group had the highest neutralization titer (only 2.7-fold lower than the HSV-2 0ΔNLS group from the first study [p = 0.011]). Substantially reducing the dose of gD DNA, whether by itself or by combining it with other glycoproteins, also reduced the neutralization titers (Fig. 9C). For instance, the neutralization titers of the 2 μg gD group and the group receiving all seven glycoproteins were significantly lower than that of the 14 μg gD group (p = 0.032 and p = 0.016, respectively). The neutralization titer of the gD+gE+gI group was also significantly lower than that of the 14 μg gD group (p = 0.016). The differences between the neutralization titers between the gD+gB, gD+gC, and gD+gH+gL groups and the 14 μg gD group did not reach statistical significance. Importantly, these results indicate that the neutralization titer did not strictly track with protection, since the groups displaying the best protection (i.e., the gD+gC and gD+gH+gL groups) had lower neutralization titers than the gD-alone group. By contrast, the neutralization titers tracked with the dose of the gD plasmid delivered.
Materials and Methods
Mice
For studies involving HSV-2 challenges, mice were housed at the BIOQUAL animal facility, and all procedures performed on the mice were performed at this facility. BIOQUAL's facility is AAALAC International accredited, USDA Registered and has an Assurance from the Office of Laboratory Animal Welfare (formerly OPRR) OLAW Assurance No. A3086-01. The 6- to 8-week-old BALB/c mice were purchased from Harlan (Frederick, MD). The studies were approved by the BIOQUAL Animal Care and Use Committee in October 2012, and they were assigned Protocol No. 12-3465-60. Mice were handled in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
For the repeat of study 2 (data shown in Fig. 9), the mice were housed at the vivarium of the Department of Comparative Medicine of New York Medical College. This facility is AAALAC International accredited, USDA Registered and has an Assurance from the Office of Laboratory Animal Welfare (formerly OPRR) OLAW Assurance No. A3362-1. The Comparative Medicine Department also complies with all the policies promulgated in the “Guide for the Care and Use of Laboratory Animals” (National Academy of Sciences, 1996).
Cells and viruses
Vero cells, 293 cells, and U2OS cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and High Five™ insect cells were obtained from Invitrogen Corporation (Carlsbad, CA). The ICP0-complementing L7 cell line (74) was kindly provided by Neal Deluca (University of Pittsburgh). Cell lines were propagated in Dulbecco's modified Eagle's medium (DMEM) that was supplemented with 5–10% fetal bovine serum (FBS) and antibiotics. The HSV-2 recombinant virus used in this study was derivative of HSV-2 MS (ATCC). HSV-2 viruses were propagated in U2OS cells at 34°C after inoculation with a multiplicity of infection of 0.01 pfu per cell. For both wild-type HSV-2 and HSV-2 0ΔNLS viruses, viral stocks were generated that were concentrated 10-fold by ultracentrifugation to achieve a minimum titer of 3 × 107 pfu/mL. More detailed methods used to construct and characterize HSV-2 recombinant viruses used in this study are provided in Halford et al. (44). An HSV-2 gD-expressing baculovirus was used to purify the gD306t protein (66) (see the section Purification of gD-2306t antigen), and was generously provided by Dr. Gary Cohen and Dr. Roslyn Eisenberg (University of Pennsylvania).
Purification of gD-2306t antigen
The gD306t protein engineered by Nicola et al. possesses an N-terminal honeybee melittin secretion signal in place of the gD leader peptide, followed by amino acids 1–306 of the mature gD protein and a C-terminal His6 affinity-purification tag (66). The gD306t protein was isolated from a flask containing 2 × 108 High Five insect cells that had been inoculated for 48 h with 2 pfu per cell of gD306t-expressing baculovirus with shaking at 27°C. Baculovirus-infected cells were removed by centrifugation, and secreted gD306t protein was purified from supernatants by dialysis against an excess of 20 mM Tris pH 8.0, 300 mM NaCl, and 10% glycerol overnight. Imidazole was added to the dialyzed supernatant to a concentration of 10 mM before affinity purification on a HisTrap™ HP column (GE Healthcare Biosciences, Piscataway, NJ) using an ÄKTApurifier™ fast-performance liquid chromatography system (GE Healthcare Biosciences). The gD306t protein was eluted from the column with 300 mM imidazole, and purity was verified at >90% by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining. Aliquots of gD306t were stored at −80°C until use.
Vaccinations with the gD protein subunit vaccine and the HSV-2 0ΔNLS vaccine
The gD subunit vaccine was prepared by combining purified gD306t with an equal volume of Imject alum adjuvant (Thermo Scientific, Rockford, IL) to achieve a protein concentration of 50 ng per μL. MPL (Avanti Polar Biolipids, Alabaster, AL) was added to a concentration of 200 ng per μL. The vaccines were stored at 4°C until vaccination the following day. Before immunization, mice were anesthetized by intraperitoneal (i.p.) administration of xylazine (7 mg/kg) and ketamine (100 mg/kg). Groups of 10 BALB/c mice were injected in their right, rear footpads with 50 μL of this formulation on day 0 such that mice were immunized with 2.5 μg of gD306t protein and 10 μg of MPL. These doses of gD306t were modeled after the gD-2 vaccine-challenge studies of Bourne et al. (16,17). Mice received an equivalent injection in their left rear footpads on day 21. HSV-2 0ΔNLS-immunized mice were injected in the footpads on days 0 and 21 as described earlier for the gD subunit vaccine, but they were immunized with 50 μL of purified HSV-2 0ΔNLS containing 1 × 106 pfu.
Synthesis of DNA vaccines
The amino acid sequences of HSV-2 gB, gD, gC, gE, gH, gI, and gL (PubMed accession No. NC_001798.1) were modified to replace their native signal sequences with the human CD5 leader sequence (MPMGSLQPLATLYLLGMLVASCLG). The amino acid sequences of HSV-2 VP5 (NC_001798), ICP8 (NC_001798), and ICP10 (NC_001798) were modified to place a tissue plasminogen activator leader sequence (MDAMKRGLCCVLLLCGAVFVSPSQEIHARFRRGAR) in front of the coding sequence to facilitate secretion. DNA sequences based on these amino acid sequences were codon and RNA optimized for expression in human and mouse cells by GenScript (Piscataway, NJ). Kozak sequences were placed upstream of the start codons, and 5′ Eco-R1 and 3′ Xho-1 site sequences were included to facilitate subcloning into the pPBS DNA vaccine plasmid (formerly named WLV-001). This plasmid has been used in numerous mouse and macaque studies and human clinical trials (32 –34,65,91). Subcloning of the DNA inserts was performed by using standard procedures utilizing the Eco-R1 and Xho-1 sites. Expression of each of the glycoproteins was verified by Flow Cytometry by positive staining using anti-HSV-2 0ΔNLS pooled sera (Fig. 5). The expression of VP5, ICP8, and ICP10 was verified by Western blots. Vaccination-grade plasmid DNA preparations of each of these plasmids and the pPBS-mIL-12 plasmid were made by Puresyn, Inc. (Malvern, PA). Endotoxin levels in the DNA preparations were <100 EU/mg.
Vaccinations with DNA vaccines
For the first study, a low-dose prime/high-dose boost approach was utilized where the priming immunizations were delivered i.m. without EP, whereas the booster immunizations were delivered i.m. with EP. Although physically less DNA was delivered during the booster immunizations, the boosters are still considered “high-dose,” because EP enhances plasmid uptake/expression in immunized muscle tissue by at least 100-fold (2,10,12,46,57,58,71,76,77,89). This regimen has been shown to produce better quality immune responses compared with a standard regimen where both immunizations are delivered by using EP (20). Immediately before all DNA vaccinations, mice were anesthetized by i.p. administration of xylazine (7 mg/kg) and ketamine (100 mg/kg). For the priming immunizations, mice received 50 μg total of vaccine plasmid DNA (equal amounts of each antigen plasmid when more than one antigen was delivered) and 25 μg of pPBS-mIL-12 plasmid DNA in a total volume of 100 μL of isotonic citrate buffer (29.3 mM sodium citrate, 0.67 mM citric acid, 150 mM NaCl and 0.34 mM EDTA, pH 6.4–6.7). The injection volume was split, and 50 μL was injected into right and left hamstring muscles of the hind legs. For the booster immunizations on day 21, mice received 10 μg total of antigen-expressing plasmid DNA and 0.5 μg of pPBS-mIL-12 plasmid DNA in a total volume of 50 μL of isotonic citrate buffer injected into the hamstring muscles of the right hind leg. Immediately after the i.m. inoculations, the injection sites were electroporated with four pulses of 100 V lasting 50 msec each with 200 msec intervals between pulses. EP was carried out by using a BTX model 830 EP generator (BTX, Holliston, MA) that was equipped with a two-needle gene probe.
For the second study, both the prime and boost immunizations were delivered i.m. with EP. All immunizations contained 14 μg total of glycoprotein plasmid DNA (equal amounts when more than one glycoprotein was given) and 0.5 μg of pPBS-mIL-12 plasmid DNA in a total volume of 50 μL of isotonic citrate buffer injected into the hamstring muscles. The priming immunizations were administered to the right hind leg, and the booster immunizations were delivered into the left hind leg. Immediately after the i.m. inoculations, the injection sites were electroporated with four pulses of 100 V lasting 50 msec each with 200 msec intervals between pulses using a BTX model 830 electroporation generator equipped with a two-needle gene probe. For the repeat of the second study, mice were vaccinated in exactly the same way as in study 2 by the same researcher.
Challenge of mice with HSV-2 in the vagina
Immunized or mock-immunized control mice were pre-treated 7 and 3 days before inoculation (days 74 and 78 post the first immunization) for the first study or 8 and 4 days before inoculation for the second study (timing was altered because of a snow storm) with 2 mg medoxyprogesterone (Depo-Provera®;, Pfizer, Inc., New York, NY) to increase the efficiency of vaginal infection (67). Immediately before HSV-2 challenge, mice were anesthetized by i.p. administration of xylazine (7 mg/kg) and ketamine (100 mg/kg). The vagina was then cleared of mucus by briefly introducing the cotton end of a cotton-tipped applicator into the vagina. On removal of the cotton swab, a pipettor was used to deliver 20 μL of complete DMEM containing 25,000 pfu/μL of HSV-2 MS virus (500,000 pfu total) into the vaginal vault.
Measurement of infectious HSV-2 titers in the vaginal mucosa
Viral titers in the vaginal secretions of mice were determined at the indicated times after inoculation by inserting a cotton-tipped applicator into the vaginal vault, and transferring the tip into 0.4 mL complete DMEM. Viral titers were determined by a 96-well plate plaque assay on Vero cells that were cultured in complete DMEM containing 0.5% methylcellulose. After 2 days of incubation in each plaque assay, cell monolayers were stained with a solution of 20% methanol and 0.2% crystal violet, and plaques were counted.
HSV-2 0ΔNLS antiserum
Ten BALB/c mice were immunized on days 0 and 30 with HSV-2 0ΔNLS, and serum was harvested on day 60 as described (43). Equal volumes of serum from the 10 individual mice were pooled. The pooled antiserum was found to react with un-transfected human 239 cells by Flow Cytometry (see the section 293 cell transfections and flow cytometry). This reactivity was caused by the HSV-2 0ΔNLS virus being grown in human cells and therefore incorporating human cell surface proteins into virion membranes. To remove the anti-human reactive antibodies from the HSV-2 0ΔNLS antiserum, 200 μL of pooled HSV-2 0ΔNLS antiserum was diluted 1:5 with sterile phosphate-buffered saline (PBS). The diluted antiserum was incubated four successive times with 10 × 106 293 cells for 10 min each at room temperature. After each incubation, the cells were pelleted by centrifugation and the cleared antiserum was removed. After this procedure, the antiserum had no detectable reactivity with un-transfected or mock-transfected 293 cells by Flow Cytometry as (see the section 293 cell transfections and flow cytometry) (data not shown).
293 Cell transfections and flow cytometry
Half a million 293 cells were plated in individual wells of six-well tissue culture plates (Corning® Costar). The next day, individual wells of cells were transfected with 1 μg of the indicated glycoprotein-expressing plasmids or an empty plasmid as a mock control using Fugene Transfection Reagent (Promega, Madison, WI). Twenty-four hours after transfection, cells were removed from the plates by using a non-enzymatic cell dissociation buffer (CellGro, Manassas, VA) and washed twice with PBS containing 1% FBS. The cells were then incubated with a 1:50 dilution of 293 cell-adsorbed HSV-2 0ΔNLS antiserum (see the section Purification of gD-2306t antigen) for 15 min at room temperature. The cells were then washed twice with 2 mL of PBS containing 1% FBS and then further incubated with a 1:100 dilution of a rabbit anti-mouse FITC-conjugated secondary antibody (Southern Biotech, Birmingham, AL) for 15 min at room temperature. The cells were then washed twice with 2 mL of PBS containing 1% FBS and then resuspended in 500 μL of PBS containing 1% FBS. The cells were then immediately run on an FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA). Data were analyzed by using FlowJo software (TreeStar, Ashland, OR).
Anti-gD ELISA
Capture ELISAs were used to determine gD-specific Ab titers in the sera of mice. Briefly, 96-well high-binding microtiter plates (Nunc, Rochester, NY) were coated with purified gD-2 (60 μL at 2 μg/mL) (see the section Purification of gD-2306t antigen) (100 μL/well) overnight at 4°C. Plates were washed three times with Tris buffered saline (TBS), then blocked with (300 μL/well) Blotto (4% dried milk in TBS) at room temperature for 30 min. Serially diluted sera were added to the wells, incubated at room temperature for 1 h, and washed three times with TBS. Horseradish peroxidase-conjugated goat anti-mouse IgG, IgG1, or IgG2a (Kirkegaard & Perry, Gaithersburg, MD) diluted 1:1,000 in Blotto were added (100 μL/well) and incubated at room temperature for 1 h. The plates were washed three times with TBS before adding SureBlue TMB peroxidase substrate (100 μL/well) and incubating for 3–5 min. The reactions were stopped by adding (50 μL/well) of 1 N H2SO4. Absorbance was read at 450 nm by using a Beckman Coulter AD 200 plate reader. Half-maximal serum binding titers were calculated by using SigmaPlot v13 software.
Anti-gD ELISpot assays
Ninety six-well flat-bottom ELISpot plates (Millipore, Bedford, MA) were coated overnight at 4°C with an anti-mouse interferon (IFN)-γ monoclonal antibody (BD-Pharmingen, San Jose, CA) at a concentration of 10 μg/mL, after which the plates were washed three times and then blocked for 2 h with PBS containing 5% heat-inactivated FBS. Mouse splenocytes were resuspended in complete RPMI 1640 medium containing either medium alone, 50 mg/mL PHA-M (Sigma), or a peptide pool (15mers overlapping by 11 amino acids; 1 μM each final peptide concentration) spanning HSV-2 gD. Input cell numbers were 4 × 105 cells/well, and they were assayed in duplicate wells. Cells were incubated for 22–24 h at 37°C, then removed from the ELISpot plates by first washing with de-ionized water, next washing six times with PBS containing 0.25% Tween-20, and finally washing an additional three times with PBS. Plates were treated with a biotinylated anti-mouse IFN-γ detection antibody (0.5 μg/well; BD-Pharmingen) and incubated at room temperature for 2 h. ELISpot plates were washed 10 times with PBS containing 0.25% Tween-20, treated with 100 μL per well of streptavidin-horseradish peroxidase conjugate (BD-Pharmingen) diluted 1:100, and incubated for an additional 1 h at room temperature. Unbound conjugate was removed by rinsing the plate 10 times with PBS containing 0.25% Tween-20. Chromogenic substrate (100 μL/well; AEC chromagen; BD-Pharmingen) was then added for 3–5 min before being rinsed away with water, after which the plates were air-dried and the resulting spots were counted by using an Immunospot Reader (CTL, Inc., Cleveland, OH). Peptide-specific ELISpot responses were considered positive if the response (minus media background) was greater than threefold above the media response and ≥50 Spot Forming Cells (SFC)/106 cells.
Neutralization assays
Two microliters of each serum sample was added to a single well in the top row of a microtiter plate containing 91 μL of complete DMEM to achieve an initial 1:46 dilution. Serial 0.33-log dilutions were achieved by serial transfer of 43 μL into 50 μL diluent (final volume = 93 μL) from the top to the bottom of the plate. A virus-complement mixture was created by diluting guinea pig complement (Rockland Immunochemicals, Gilbertsville, PA) 1:50 in complete DMEM and adding HSV-2 MS to a concentration of 3,500 pfu per mL. The HSV-2 neutralization assay was initiated by combining 50 μL of the virus-complement mixture with each serum dilution (50 μL) and incubating at 37°C. After 2 h, 100 μL of a suspension containing 4 × 106 Vero cells per mL was added to each well, and microtiter plates were incubated for 48 h to allow HSV-2 plaques to form. Cell monolayers were fixed and stained with a 20% methanol, 0.1% crystal violet solution. The HSV-2 neutralizing titer of each serum sample was considered the reciprocal of the largest serum dilution that reduced HSV-2's cytopathic effect in Vero cell monolayers by at least 50%.
Statistical analysis of results
Viral titers were transformed by adding a value of 1 such that all data could be analyzed on a logarithmic scale. The error bars in the figures are the standard errors of the means (SEMs). The SEMs were calculated by dividing the standard deviations of the means (determined using MicroSoft Excel software) by the square root of the number of animals in the group. The significances of differences between two treatment groups for viral shedding, weight loss, antibody titers, or neutralization titers were compared by t-tests if both the data sets were normally distributed or by Mann–Whitney Rank Sum Tests if both the data sets were not normally distributed. The analyses were performed by using SigmaPlot version 13 software by Systat Software, Inc. (San Jose, CA). A p-value ≤0.05 indicates that there is a significant difference in the immune measure between groups. The significance of differences in survival frequency between immunization groups was determined by Fisher's Exact Test. The significance of differences in percent survival after HSV-2 MS challenge of 0ΔNLS-immunized mice and gD-2-immunized mice was compared by a two-sided Student's t-test.
Discussion
We first compared the protective efficacy of an IL-12-adjuvanted gD DNA vaccine delivered by using a low-dose prime/high-dose boost regimen with a recombinant gD subunit vaccine adjuvanted with alum and MPL and with the highly protective live attenuated vaccine HSV-2 0ΔNLS (43,44). Consistent with what was found in previous published studies (43), the HSV-2 0ΔNLS vaccine was dramatically more protective than the subunit vaccine in terms of survival from challenge, reducing both weight loss and virus shedding post-challenge (Figs. 1 –3). By contrast to the gD subunit vaccine, the IL-12-adjuvanted gD DNA vaccine provided similar protection from death and weight loss post-challenge as the HSV-2 0ΔNLS vaccine (Figs. 1 and 2), but the reduction in virus shedding provided by the gD DNA vaccine was more similar to that provided by the subunit vaccine than it was to that provided by HSV-2 0ΔNLS (Fig. 1). These results indicate that the gD-expressing DNA vaccine is vastly superior to the subunit vaccine for protection from death and weight loss post-challenge but only marginally superior for reducing virus shedding.
The gD subunit vaccine elicited mostly Th2-associated anti-gD IgG1 isotype antibodies, and very little Th1-associated IgG2a antibodies that are associated with Fc-mediated effector functions (Fig. 4A). This result is consistent with the hypothesis that gD subunit vaccines are poor inducers of antibody responses that are capable of effector functions even when they are delivered with potent adjuvants. By contrast, the IL-12-adjuvanted gD DNA vaccine elicited antibody responses that were skewed toward the IgG2a isotype (Fig. 4A). Further, the gD subunit vaccine elicited substantially lower neutralizing titers than the HSV-2 0ΔNLS or gD DNA vaccines (Fig. 4C). This result may be a reflection of the fact that the neutralization assay performed is dependent on the addition of complement (38). Therefore, at least some of the neutralization measured in this assay likely requires antibodies that are capable of fixing complement (i.e., antibodies with Fc-mediated effector functions such as IgG2A).
Since the total magnitudes of the IgG antibodies evoked by the two different gD vaccines were not significantly different, the IgG2a skewed humoral response elicited by the gD DNA vaccine may be responsible for its increased efficacy over the gD subunit vaccine. It is possible, however, that other factors such as improved cellular immune responses or improved mucosal responses also contributed to the improved efficacy of the gD DNA vaccine. Arguing against these possibilities are one that we are unaware of data indicating that DNA vaccines delivered i.m. provide enhanced mucosal immunity compared with other delivery regimens, and two that gD is not considered an important antigen for generating useful cellular immune responses (47,69,70). In addition, DNA vaccination with the intracellular HSV-2 antigens VP5, ICP8, or ICP10 (which likely evoke robust cellular immune responses) provided little protection in this high-dose HSV-2 challenge model and did not substantially improve the protection afforded by the gD DNA (Figs. 1 –3), further arguing against cellular immune responses playing an important role in protection in this high-dose challenge model.
There are also several possible reasons for the superiority of the HSV-2 0ΔNLS vaccine over the gD DNA vaccine, including increased antigenic breadth, increased cellular immune responses, and increased mucosal immune responses. We sought to increase the antigenic breadth of our DNA vaccine by first combining gD with humoral-immunodominant intracellular viral antigens. Surprisingly, VP5, ICP8, and ICP10 (40) did not provide significant protection in this high-dose HSV-2 challenge model (Figs. 1 –3). It should be noted, however, that the lack of efficacy displayed by these antigens may be an artifact of the high-dose challenge model used. In this regard, it is likely that the massive challenge dose (∼2,000 times the LD50) overwhelms the ability of cellular immune responses to combat the infection. Therefore, our results may not be reflective of the true potential of these proteins as vaccine antigens. Unfortunately, such a large challenge dose is necessary to detect differences in glycoprotein-based vaccines that likely offer protection through humoral means.
We next sought to increase the antigenic breadth of our DNA vaccine by including other potentially relevant glycoprotein antigens. Seven glycoproteins (gB, gC, gD, the gE/gI heterodimer, and the gH/gL heterodimer) are involved in HSV-2 cell entry and cell-to-cell spread [reviewed in Akhtar and Shukla (3), Connolly et al. (28), and Eisenberg et al.(35)]. We, therefore, determined the extent to which these six other glycoproteins could enhance the protective efficacy of a gD-based DNA vaccine.
Each of the glycoprotein antigens enhanced the efficacy of the gD DNA vaccine in terms of survival, reduced weight loss/increased weight regain, and/or reduced virus shedding (Figs. 6 –8). Compared with gD alone, the addition of gC or the gH/gL heterodimer provided the best improvements in survival, weight loss/weight regain, and reduced virus shedding. Our results, therefore, support the previously suggested notion that co-targeting gC and gD makes for a good vaccine strategy (7 –9). This other group is targeting gC based on the hypothesis that antibodies that inhibit the immune evasive function of gC will enhance vaccine efficacy (8). The results of our unbiased glycoprotein screen support this hypothesis. The addition of gB also provided improvements in survival, and weight loss/weight regain, but did not reduce virus shedding. It should be noted, however, that the performance of gB in our studies may have been suboptimal due to low expression levels of the gB-expressing plasmid (Fig. 5). We have since improved the expression of our gB plasmid by genetically removing the cytoplasmic tail from gB as described (6) (not shown).
The gE/gI heterodimer did not enhance survival or reduce viral shedding, but it did provide for the best weight regain (Figs. 6 –8). This could reflect an effect of the anti-gE/gI antibodies on preventing virus spreading, allowing the mice to regain normal behaviors and normal appetites sooner. Vaccination with all seven of the glycoproteins provided an enhancement in survival and reduced late (day 5) virus shedding but did not improve weight loss or regain (Figs. 6 –8). An important caveat is that the total glycoprotein doses of DNA administered during the vaccinations were kept the same. Therefore, the mice receiving all seven glycoproteins received sevenfold less of each plasmid than the mice receiving the gD plasmid alone. This is important, because we also found that reducing the dose of the gD plasmid delivered caused a corresponding drop in the magnitudes of the anti-gD humoral and cellular immune responses (Fig. 9). We, therefore, believe that it is likely that the increase in antigenic breadth provided by delivering multiple glycoprotein antigens was either partially or completely offset by the corresponding reductions in magnitudes of the antibody responses raised against these antigens. In other words, broad responses of suboptimal magnitude may be of limited benefit over optimal responses that focused on single antigens.
Although HSV-2 0ΔNLS possesses numerous antigens and evokes antibody responses against a large number of these antigens [Fig. 5 and also see reference (40)], the magnitudes of the antibody responses evoked against these antigens by HSV-2 0ΔNLS remain high (Fig. 4A). The higher magnitudes of the antibody responses to the individual glycoprotein immunogens evoked by HSV-2 0ΔNLS compared with the multi-valent DNA vaccines are likely partially or fully responsible for the greater efficacy of HSV-2 0ΔNLS in terms of virus shedding (Fig. 1). It may be possible to increase the efficacy of the multi-valent DNA vaccines by increasing the magnitude of the responses to each of the multiple antigens in the DNA vaccine by increasing the total DNA dose delivered, increasing the number of booster vaccinations and/or by employing a protein boost approach (39).
Although the results of our studies are promising, we are certainly not the first to demonstrate that DNA vaccines targeting HSV-2 antigens can be protective. In fact, studies demonstrating the effectiveness of DNA as a delivery modality for HSV-2 vaccines, including those targeting gD, go back nearly two decades (79 –82). We are also not the first to demonstrate that the IL-12 adjuvant enhances the effects of HSV-2 DNA vaccines (81,82), and we are not the first to demonstrate that targeting glycoproteins other than gD can be protective alone or when combined with gD (5,8,19,30,61,64,68). We do, however, believe that our current studies are the first to evaluate HSV-2 glycoprotein-based DNA vaccines delivered with IL-12 by EP, which enhances antigen expression at least 100-fold (2,10,12,46, 57,58,71,76,77,89). We also believe that our results are the first to demonstrate a dramatic increase in protection from a high-dose HSV-2 challenge for a gD-based DNA vaccine compared with an adjuvanted gD-based subunit vaccine.
All said, our data indicate that DNA vaccination with the IL-12 adjuvant may offer an improved modality for delivering HSV-2 vaccines, especially when the low-dose prime/high-dose boost approach is used. Our data also indicate that each of the seven glycoproteins involved in HSV-2 cell entry and cell-to-cell spread can contribute to the efficacy of a DNA-based HSV-2 vaccine in the mouse model. Finally, our data indicate that a multi-glycoprotein-expressing DNA could approach the efficacy of live attenuated HSV-2 vaccines if the magnitude and perhaps durability of the humoral immune responses to the individual components of the vaccine can be increased.
Footnotes
Author Disclosure Statement
No competing financial interests exist.