
Abstract
The aim of this study was to design a multiepitope universal vaccine for major human papillomavirus (HPV) structural proteins, L1/L2, by bioinformatics models. For this purpose, we predicted the most probable immunogenic epitopes of L1 and L2 from common high-risk HPV 16, 18, 31, and 45 beside high prevalent type 6 and 11 based on BCPREDS defaulted model, while solvent accessibility of structure was extrapolated. The three-dimensional molecular model of L1 protein was constructed by Swiss Model server, whereas sequence alignment provided model for prediction of L2 protein epitopes. After that, N-glycosylation sites were excluded from estimated epitope regions. Then, by other bioinformatics analyses, 20 epitopes were selected and fused in tandem repeats, reverse translated, and codon optimized to relevant sequence. The final protein parameters such as antigenicity were analyzed by protean program. Evaluation of new recombinant protein sequence indicated a molecular weight of 41.8 kDa with 400 amino acids beside positive charge. The computed isoelectric point (pI) value indicated the acidic nature of final product. The aliphatic index showed low thermal stability of this construct and the Grand Average Hydropathicity value was negative (−0.494). Analyzed plot showed that major parts of new protein construct had hydrophilic property, thus harboring antigenic potency. After all, sequence of final construct reverse translated to DNA and this codon-optimized sequence showed Codon Adaptation Index (CAI) of >0.8 for expression in Escherichia coli. Finally, this sequence ligated into pET28a bacterial expression vector. The new recombinant proteins harboring 20 B cell epitope seem to be suitable antigens based on computational methods as a universal vaccine candidate for HPVs.
Introduction
H
The papillomavirus genome is ∼7.9–8 kb long and contains three main regions: a noncoding region (NCR), termed also as long-control region (LCR) or upstream regulatory region (URR), an early (E) region of open reading frames (ORFs), and a late (L) region of ORFs. The circular papillomavirus genome encodes roughly eight ORFs from a single DNA strand (18), which is responsible for viral replication, maintenance, and cell transformation. These gene products have been classified into six regulatory (E1-E6) and two structural proteins (L1 and L2) (3). An extensive body of researches has demonstrated that E1 and E2 are involved in viral transcription and replication; E5, E6, and E7 act as oncoproteins, while L1 and L2 contribute to the formation of the capsid (new). The viral icosahedral capsid is built up from 72 identical capsomers that comprised five L1 and one L2 molecule. The exposed L1 and L2 proteins have been identified as competent targets for prophylactic vaccination as they are expressed during early infection (13). The L1 is the major capsid protein with molecular weight of 55 kDa, being particularly abundant and representing 80% of the capsid proteins (ratio L1/L2 10:1). L2 protein is about 500 amino acids long with molecular weight of 64–78 kDa and representing ∼20% of the capsid proteins, thus being poorly represented on the surface of HPV virions (18).
Even providing an effective vaccine did not control the rate of cervical cancer in population of developing countries. Different kinds of vaccination strategies have been developed for HPV; among them, peptide-based and virus-like particle vaccines have considerable value (13). The FDA-approved HPV vaccines designated as Gardasil and Cervarix, composed of self-assembling L1 moieties in virus-like particle structures, have entered into market in 2006 and 2009, respectively (5). The Gardasil harbors a mix of HPV-16/18/6/11 L1-VLP, while Cervarix composed HPV-16/18 VLPs. Despite ideal results that were imagined for these approved vaccines, some drawback looks inevitable. One considerable shortcoming is their limited serotype coverage (9). The induced humoral immunity of these vaccines is primarily type specific, so this level of protection will not support complete protection from other oncogenic types. In sum, with respect to vaccine cost, restricted type coverage, and person-dependent efficacy of Gardasil and Cervarix for high-risk serotypes, designing a new preventive/therapeutic vaccine with extended coverage potency is an essential task (2,14).
The epitope-based vaccines can be designed for humoral and cellular stimulation and is very safe approach when compared to other strategies. B lymphocytes play a crucial role in the humoral immunity of adaptive immune system. The distinguishing of B cell epitopes as immunogenic regions within a target protein is the goal of some immunologic assays. Similar to cellular epitopes, B cell epitopes can play a vital role in the development of peptide vaccine (24). Identification of B cell epitopes reactive to HPV L1-derived peptides performed in animal model several times with the aims of designing effective epitope-based vaccine (7). The experimental techniques are time-consuming and labor-intensive, therefore several in silico methodologies are being created to distinguish epitopes of protein (6). On another side, computational techniques offer fast, easy, reliable, and cost-effective approaches for predicting B cell epitopes. Using bioinformatics tools, scientists can analyze the protein of interest for extraction of epitopes rather than potential binding sites in epitope-based vaccines (6,7). Furthermore, improved reliability of computational models in the prediction of desired B cell epitope will surely facilitate a preexperimental step of vaccine development.
Development of a new-generation peptide-based vaccine covering more carcinogenic serotypes may get rid of limited coverage of current HPV vaccines. Others also designed VLP-based vaccine by engineering of recombinant L1 VLP protein containing major preS1 epitopes of hepatitis B virus using bioinformatics analysis (10). Also, in silico analysis was applied to the development of peptide vaccine targeting different HPV proteins (11). They surveyed along 20 L1 sequence, and afterward, three conserved B cell peptides were extracted for next experimental assay. Recently, in an attempt to design multiepitope therapeutic vaccine against cancer, one group employed the benefit of computational models (15).
In this study, a multiepitope vaccine against major HPV structural proteins L1/L2 was designed by immunoinformatics models considering the coverage potency for HPV 16, 18, 31, 45, 6, and 11 subtypes as a universal vaccine. These proteins are considered to be immunogenic for vaccines design. For this purpose, to obtain surface and conserve B cell epitopes in silico by a convenient computational method, we extended informatics strategies on different virus genes and tried to identify consensus B cell epitopes suitable for prophylactic regimen. To our knowledge, reports of B cell epitope prediction for L1 and L2 of HPVs is limited; so we predicted the most probable immunogenic regions of L1 and L2 among high-risk HPV 16, 18, 31, 6, and 45 rather than type 11 serotypes based on BCPREDS defaulted model, and solvent accessibility of structure was extrapolated. For determination of solvent accessibility, the precise structure of protein is necessary. The crystal structure of L1 and L2 proteins was not determined. So we constructed a three-dimensional (3D) molecular model of the L1 by Swiss Model server, but this server was unable to provide model of L2 protein. Finally, based on best estimated epitope regions extracted from molecular modeling analysis, N-glycosylated sites, codon optimization, and other bioinformatics analyses, B cell epitope relevant to L1 and L2 protein was extracted and modified and then presented to new HPV constructs for the next experimental assessment.
Materials and Methods
Protein sequences collection
Protein reference sequences of L1 and L2 from common/high-risk HPVs, including type 6, 11, 16, 18, 31, and 45, are retrieved from
HPV, human papillomavirus.
B cell epitope prediction
For the prediction of epitope regions in L1 and L2 proteins, the BCPREDS server (
Solvent accessibility of epitopes
After selection of epitopes, these epitopes were refined by the degree of solvent accessibility. Each residue at the surface of a protein can potentially be touched by water, and the area of an atom on the surface that can be touched by water is called the accessible molecular surface or solvent-exposed area. So it is advisable to select epitopes with higher degree of solvent accessibility. For fulfillment it, the structure of the L1 and L2 proteins was needs. The crystal structures of L1 and L2 protein had not been determined before. For obtaining a 3D structure model of L1 proteins, the crystal structure of these proteins was used as PDB template in Swiss Model Alignment interface protein modeling server (10). For L1 HPV 11, the crystal structure of L1 HPV 11 (PDB code: 2r5kD, Uniprot Entries Accession P04012); for L1 HPV 16, the crystal structure of L1 HPV 16 (PDB code: 1dzlA, Uniprot Entries Accession P03101); for L1 HPV 18, the crystal structure of L1 HPV 18 (PDB code: 2r5iG, Uniprot Entries Accession P06794); for L1 HPV 31, the crystal structure of L1 HPV 16 (PDB code: 1dzlA, Uniprot Entries Accession P03101); and for L1 HPV 45, the crystal structure of L1 HPV 18 (PDB code: 2r5iG, Uniprot Entries Accession P06794) was used. Despite submission sequence of L2 proteins, 3D structure models of L2 were not obtained. The fit between the 3D structures of models was evaluated in SWISS-PDB Viewer by calculating the root mean square deviation after iterative fitting. After preparation of structures, the solvent accessibility of these epitopes was studied in SPDBV software. Finally, the epitopes with a high degree of solvent accessibility were selected.
N-glycosylated sites
In N-glycosylation, carbohydrate chains are attached to nitrogen of asparagine and serve various functions. N-linked glycosylation is the most common type of glycosidic bond and is important for the folding of some eukaryotic proteins and occurs in eukaryotes, but very rarely in bacteria. We designed a final expression of construct in Escherichia coli, therefore it is necessary to delete the epitopes that have N-glycosylated sites. For prediction of N-glycosylated sites, the ensemblegly-Iowa server was used. Epitopes which selected from the previous step were analyzed in the ensemblegly-Iowa server and sequences that have an N-glycosylated site were excluded in the design of final construct.
Reverse translation of first structure
After selection of best epitopes of L1 and L2 based on bioinformatics analysis, these epitopes fused to each other, and the first structure of this construct was created by the Lasergene seqbuilder server. The final construct back translated into nucleotide sequences using reverse translate tool of Sequence Manipulation Suite (
Codon optimization, rare codon determination
Codon optimization is a technique that supports the efficiency of DNA expression vectors used in DNA vaccination to achieve optimum expression of a foreign gene in the host's cell body (18). Constructs were optimized using the OPTIMIZER tool in genome server (
Design of vaccine constructs
Protein analyses such as surface probability plot, alpha and beta aliphatic region, translation of antigenic sequence, antigenicity index, and prediction of binding affinity of epitopes were measured as last check point for validity and reliability of construct by Hopp and Woods method in Expasy tools (
Results
B cell epitope prediction
By the use of BCPred method in BCPREDS server of epitope regions in L1 and L2, protein sequences were determined. A total of 71 epitopes for L1 and 70 epitopes for L2 HPV serotypes were predicted. In the next step, epitopes with high score were used as candidates for application in vaccine construct (Table 2).
Solvent accessibility of epitopes
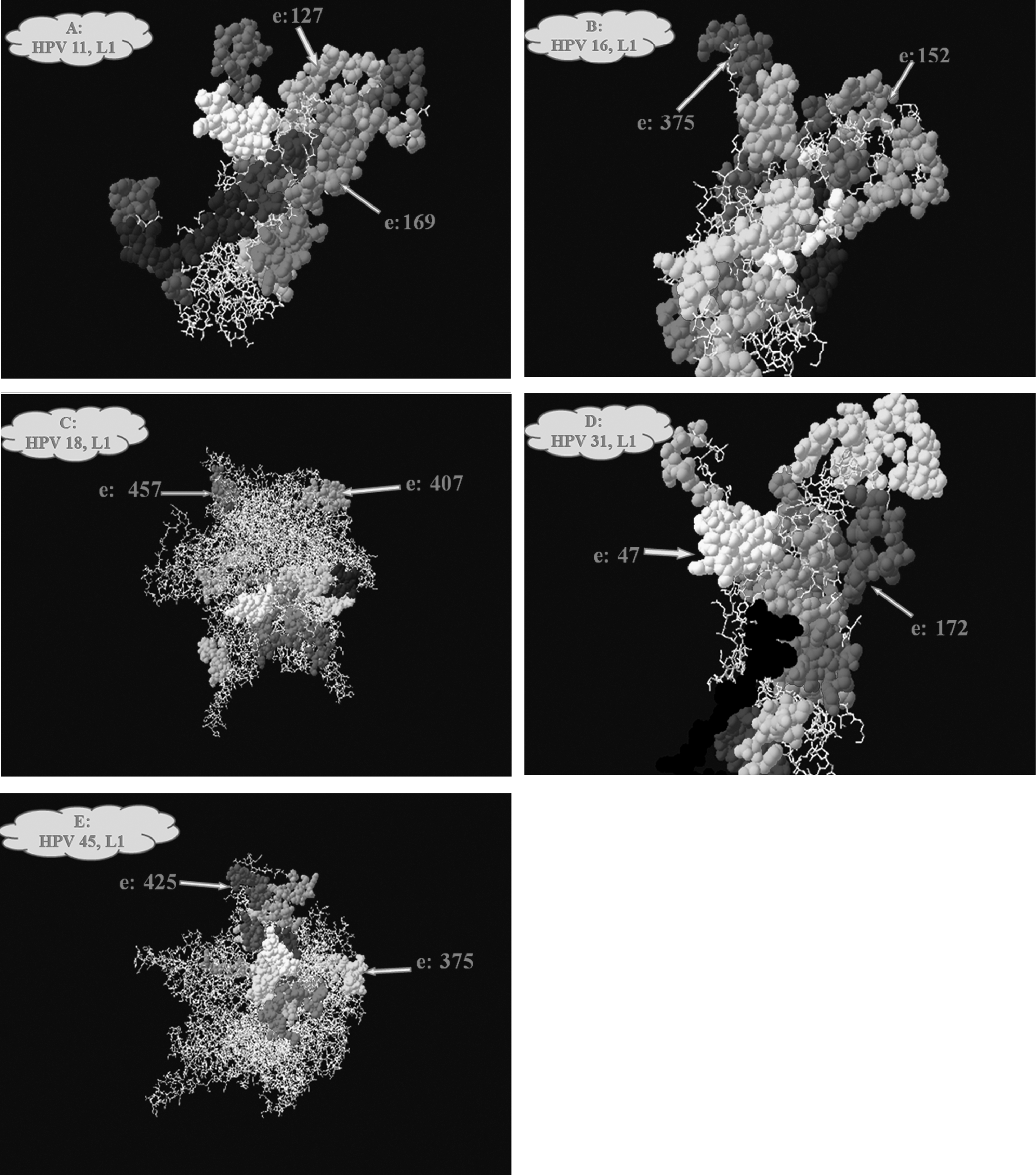
After position extraction, suggested epitopes were refined by the degree of solvent accessibility. To select epitopes with a higher degree of solvent accessibility, the structure of the L1 and L2 proteins was studied. The 3D structure models of L1 were obtained in the Swiss Model Alignment interface protein modeling server due to lack of the crystal structure for this protein. The results of homology modeling for L1 proteins from HPV types 11, 16, 18, 31, and 45 are listed in Table 3. As previously mentioned, this server was unable to model L2 protein. For further analysis of epitopes in L2 proteins, multiple sequence alignment of L2 protein from retrieved HPV 11, 16, 18, 31, and 45 serotype demonstrated that amino acid residue 11–200 have 50% homology. Therefore, extracted epitopes from this area were selected as epitopes of choice for the next step. Thereafter, the solvent accessibility of these epitopes was studied in the SPDBV software. Finally, as shown in Figure 1, the epitopes with a high degree of solvent accessibility were selected.
N-glycosylated sites
By the use of ensemblegly-Iowa server, N-glycosylation site of these epitopes was predicted. Epitopes analyzed in this server and position of asparagine that glycosylated were determined. To ensure for expression of construct in E. coli, epitopes that have an N-glycosylated site were excluded in the design of final construct. The score and potential of asparagine residue in the sequence of epitopes are demonstrated in Table 4.
The residues that accept a glycosidic bond is shown with a plus sign (A: L1 HPV 11, B: L2 HPV 11, C: L1 HPV 16, D: L2 HPV 16, E: L1 HPV 18, F: L2 HPV 18, G: L1HPV 31, H: L2 HPV 31, I: L1 HPV 45, J: L2 HPV 45).
Selecting epitopes and fusion of peptides for generating vaccine constructs
After the evaluation of epitope scores, two optimal epitopes were selected for each HPV type. L1 and L2 protein epitopes of serotype 6 and 11 were similar; therefore, we selected one epitope for both. As shown in Table 5, finally 20 epitopes were selected. These epitopes were connected to each other and the first structure of this construct was created (Fig. 2). The tandem order of them was determined just by the order of their appearance in Table 5 and they are designated as E1 to E10 for L1 protein and E11 to E20 for L2 protein.

Design of primary structure for final construct from selected epitope. Number of HPV are determined in Table 5. HPV, human papillomavirus.
Evaluation of protein sequence
The sequences of primary construct were analyzed in Expasy's ProtParam tool and results are presented in Table 6. The results indicated that molecular weight of this protein was 41.8 kDa and has 400 amino acid residues. Because the total number of positively charged residues was nearly twice that of positively charged residues, the computed isoelectric point (pI) value was below 7 (4.71), which indicates the acidic nature of final product. The aliphatic index shows that this construct has low thermal stability. The Grand Average Hydropathicity (GRAVY) value was negative (−0.494). Besides, amino acid composition revealed that most abundant amino acid residues in our construct were glutamine and leucine, respectively. The antigenicity of construct was analyzed by protean program. This plot showed that major parts of new protein construct have hydrophilic property and thus harbor antigenic potency (Fig. 3).
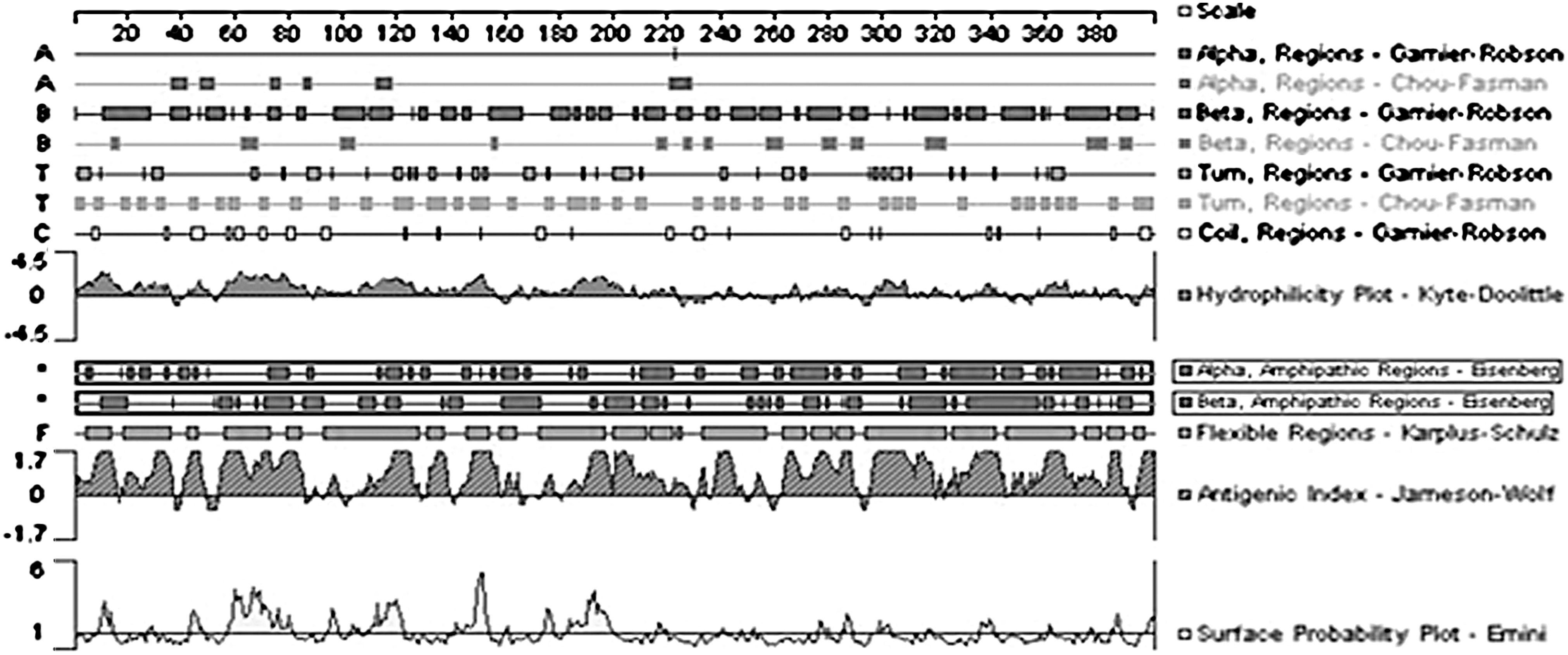
The diagram of antigenicity determined by protean software for final construct.
AI, aliphatic index; GRAVY, grand average hydropathicity; pI, theoretical isoelectric point.
Reverse translation of first structure
After confirmation of the new protein antigenicity, amino acid sequences of this construct back translated into nucleotide by sequence manipulation suite server. This process was conducted based on codon usage table, and consensus sequence derived from all the possible codons is also returned. Reverse-translated sequence was evaluated for codon optimization and rare codon to enhance gene expression level and protein solubility. For this purpose, OPTIMIZER and GenScript's servers were employed together. This information is very useful to predict that the final construct gene will express well in E. coli host. A CAI of 1.0 is considered ideal, while a CAI of >0.8 is rated as good for expression in the desired expression organism. The lower the number contain the higher chance to gene be expressed poorly. GenScript's Optimum Gene™ codon optimization tool can typically improve the sequence to reach a CAI of >0.8. The GC content of gene was 54.26%. The ideal percentage range of GC content is between 30% and 70%. Any peaks outside this range will adversely affect transcriptional and translational efficiency. The percentage of low-frequency codons (<30%) based on our target host organism is 3%. This unoptimized gene employs tandem rare codons that can reduce the efficiency of translation or even disengage the translational machinery. Results are shown in Figures 4 and 5.
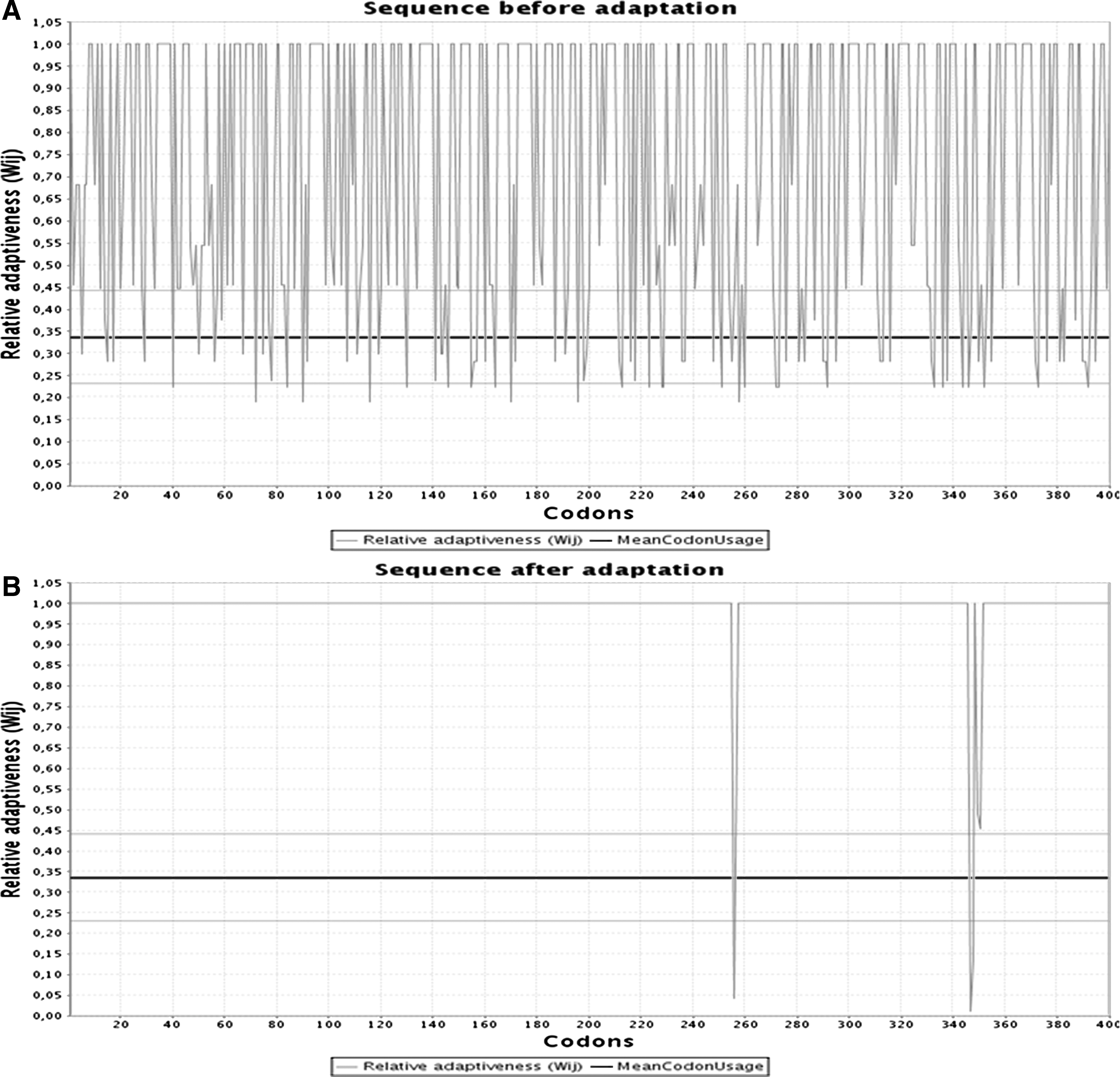
Codon optimization of final construct. Plot

Representation of reverse translation and codon optimization. This screenshot shows translation of construct and optimization (adaptation) to E. coli codons.
Discussion
So far, three prophylactic HPV vaccines, based on VLP technology, have been approved and licensed. They are highly effective and immunogenic if administered before HPV exposure, which is to say, before the commencement of an active sexual life (18). L1-based prophylactic vaccines, such as Gardasil®, Cervarix®, and Gardasil 9®, have been already licensed, while L2-based second-generation preventive vaccines are still under clinical trials. An important limitation of L1-based vaccines is that they induce type-restricted immune responses. L2-based vaccines can overcome this issue (18). The replication process of some viruses is error prone, so evolving of different strains is unavoidable; therefore, developing an effective vaccine with all serotypes coverage is nearly impossible. Although establishment of a universal vaccine for high evolving viruses such as influenza and HIV has many difficulties, some achievements have been published recently (16,23). Even providing an effective vaccine for HPV did not decrease the rate of cervical cancer in developing countries. HPV prevention remained a big concern, considering some shortages such as the presence of different genotypes and vaccine cost and availability (2,21). While around 15 high-risk HPVs were identified, current vaccines just protect humans from three types. The narrow coverage of current vaccine seems to be the most important limitation (2). The Gardasil and Cervarix harbor a mix of HPV-16/18/6/11 and HPV-16/18 L1 VLPs, respectively. Among all candidates, protein-based vaccine is a safe method for prevention and therapy (22). Development of low-cost and universal vaccine covering more carcinogenic serotypes may get rid of current HPV vaccines shortcoming (26). Theoretically, development of multiepitope-based vaccine containing common exposed sequences of HPV proteins induced wide spectrum of B cell immunity that consequently will establish potent humoral immunity. In fact, B lymphocytes play a crucial role in the humoral immunity of adaptive immune system. Similar to HLA class I and II epitopes, B cell epitopes also play a vital role in the development of protein vaccines. The B cell detects exposed parts of antigen and triggers potent humoral immune responses. The identification of exposed immunogenic regions within a protein, specifically those that elicit humoral immune response had been the goal of many studies before (2,25). Identification of B cell epitopes for HPV proteins was performed in animal models several times to design multiepitope-based vaccine (7,23). Identification of exposed motifs from virus antigens that contributed in binding to antibodies is helpful for this purpose (7,10). Immunobioinformatics analysis was applied for this kind of prediction. While experimental distinguishing of B cell epitopes is costly and time-consuming, in silico screening provided a convenient surrogate. In a previous study, Subramanian and Chinnappan (24) employed immunoinformatics tools to extract linear B cell epitope for E6 protein of high-risk HPVs. Kaushik et al. (12) by in silico analysis constructed peptide-based vaccine targeting L1 protein. In this study, we purposed a single polytopic candidate construct with six significant serotypes coverage (four high-risk HPVs and two high-rate HPV serotypes) as a new and universal vaccine candidate. Four high-risk and two low-risk HPV L1 and L2 protein sequences were evaluated for epitope predictions by the help of different immunoinformatics methods. At the end, 20 epitopes were selected as candidates for designing final construct and these peptides fused together in tandem without linker. After first epitope screening, nearly 140 epitopes were selected from 6 common HPV types. For the next step, 20 epitopes with higher degree of solvent accessibility and also higher degree of homology were selected. One of the most cited features of the epitope is hydrophilic property of surrounding sequence. This feature was first described by Novotny who calculated the solvent accessible surface area of epitopic residues (5). To select epitopes with higher degree of solvent accessibility, the 3D structure of the L1 and L2 proteins was constructed. For L2 protein, multiple alignments were performed on HPV 11, 16, 18, 31, and 45 serotypes to distinguish homology. It demonstrated that 50% homology among amino acid residue 11–200 present therefore extracted epitopes from this area selected as epitopes of choice for next step. These epitope sequences fused together in a tandem. Evaluation of new recombinant protein sequence indicated a molecular weight of 41.8 kDa with 400 amino acids. The computed isoelectric point (pI) value was 4.71, which indicates the acidic nature of final product. The aliphatic index showed that this construct has low thermal stability. Analyses of the GRAVY values showed a negative score of −0.494, indicative of suitable antigen solubility. Check of antigenicity also revealed that major parts of new protein construct have hydrophilic property and thus harbor antigenic potency. This finding also confirmed the exposure potency of almost all parts of newly constructed fusion polypeptide. Afterward, sequence of final construct was reverse translated to DNA sequence and then codon optimized. This codon-optimized sequence showed CAI of >0.8 for expression in E. coli and this score is acceptable. Finally, this sequence ligated into a pET28a bacterial expression vector through BamHI and HindIII restriction sites. Based on rational design of this construct, we expected that this construct would be a suitable candidate for universal vaccines against six common HPV types that are responsible for >90% of cervical cancer. Future generation of new highly immunogenic and effective vaccines such as L1/L2-based, capsomer-based, and pseudovirion-based vaccines can be further developed, thanks to computer-aided design and bioinformatics/computational biology (18). This kind of prediction and design by the help of bioinformatics will alleviate cost, time, labor, and space of animal experiments, especially when constructing a vaccine. Our designed construct unenviably need further experimental confirmation but at least validated as a universal derived antigen when evaluated computationally. Next, the universal vaccine will be expressed and immune responses against vaccine candidate would be evaluated in an experimental animal model.
Footnotes
Author Disclosure Statement
No competing financial interests exist. None of the work was paid for by the Iranian government or performed by its employees.