
Abstract
Infection with adenovirus is a major cause of infectious mortality in children following hematopoietic stem-cell transplantation. While adoptive transfer of epitope-specific T cells is a particularly effective therapeutic approach, there are few suitable adenoviral peptide epitopes described to date. Here, we describe the adenoviral peptide epitope FRKDVNMVL from hexon protein, and its variant FRKDVNMIL, that is restricted by human leukocyte antigen (HLA)-C*0702. Since HLA-C*0702 can be recognized by both T cells and natural killer (NK) cells, we characterized responses by both cell types. T cells specific for FRKDVNMVL were detected in peripheral blood mononuclear cells expanded from eight of ten healthy HLA-typed donors by peptide-HLA multimer staining, and could also be detected by cultured interferon γ ELISpot assays. Surprisingly, HLA-C*0702 was not downregulated during infection, in contrast to the marked downregulation of HLA-A*0201, suggesting that adenovirus cannot evade T cell responses to HLA-C*0702-restricted peptide epitopes. By contrast, NK responses were inhibited following adenoviral peptide presentation. Notably, presentation of the FRKDVNMVL peptide enhanced binding of HLA-C*0702 to the inhibitory receptor KIR2DL3 and decreased NK cytotoxic responses, suggesting that adenoviruses may use this peptide to evade NK responses. Given the immunodominance of FRKDVNMVL-specific T cell responses, apparent lack of HLA-C*0702 downregulation during infection, and the high frequency of this allotype, this peptide epitope may be particularly useful for adoptive T cell transfer therapy of adenovirus infection.
Introduction
I
During infection, adenovirus is able to evade T cell responses by downregulating the expression of human leukocyte antigen (HLA) molecules (6). More recently it has been shown that adenovirus can also evade natural killer (NK) responses by downregulating the HLA-like MICA/B molecules that activate NK responses (24,30). These effects are predominantly mediated by the adenoviral gene product E3 19K, which binds and sequesters HLA and HLA-like molecules intracellularly (31).
While downregulation of common HLA types such as HLA-A*0201 and HLA-A*0301 has been well studied, little is known about the regulation of HLA-C types during adenoviral infection (15,22). This is relevant because all HLA-C types are recognized by killer inhibitory receptors expressed on NK cells, such as KIR2DL3 for group 1 HLA-C allotypes, including HLA-C*0702 (8). Although binding of KIR is not specific for peptides presented on the relevant HLA-C allotypes, it can be influenced by residues in the C-terminal region of the peptide resulting in alterations in NK responses (8,27). Presentation of peptide epitopes on HLA-C allotypes thus allows coordinated surveillance of viral infection by specific T cells and NK cells. Such coordinated surveillance of infection may be superior to surveillance by T cells alone, which may be more susceptible to immunoevasion in the absence of NK activation.
This is particularly well described during HIV infection (21). HIV evades immune responses by mutating presented peptides not only to prevent their recognition by T cells but also to enhance binding to KIR and, therefore, to enhance inhibition of NK responses (11,12,20,33,34). Novel adenoviral peptide epitopes presented by HLA-C allotypes may therefore be relevant for adoptive T cell transfer therapy.
In the present study, we identify a peptide epitope FRKDVNMVL and its variant FRKDVNMIL, from adenovirus hexon protein that is presented on HLA-C*0702. We show that FRKDVNMVL-specific T cell responses are immunodominant, and that adenovirus surprisingly does not downregulate HLA-C*0702 expression during infection as a possible strategy to evade the T cell response. By contrast, presentation of FRKDVNMVL on HLA-C*0702 enhances binding to the inhibitory receptor KIR2DL3 and allows evasion of NK cytotoxic responses. The FRKDVNMVL peptide epitope may be useful for adoptive T cell transfer therapy of adenovirus infection.
Materials and Methods
Cells and constructs
HLA-A*0101 (from Elizabeth Jaffee, Johns Hopkins University, Baltimore, MD), HLA-B*0702 (GeneCust, Dudelange, Luxembourg), and HLA-C*0702 (from Josef Mautner, Munich, Germany) constructs were cloned into the vector pMXsIP (BioCat, Heidelberg, Germany) and expressed by retroviral transduction in K15 cells (K562 expressing membrane-bound interleukin-15, CD137 ligand, and green fluorescent protein (GFP) kindly provided by Dario Campana, St. Jude Children's Research Hospital, Memphis, TN) (17). K15 cells were also transduced with HLA-C*0702 linked to the sequences of either FRKDVNMVL or FRKDVNMIL, HLA-A*0101 linked to LTDLGQNLLY, and HLA-B*0702 linked to VPATGRTLVL in pMXsIP as previously described (5,25). Expression was verified by staining transduced cells with W6/32 pan-HLA class I antibody (eBiosciences, Frankfurt, Germany). NK-92MI cells were transduced with KIR2DL3 (GeneCust) in pMXsIP (9), and expression was verified using KIR2DL2/3 antibody (BioLegend, Aachen, Germany).
Expansion of T cells and ELISpot assays
Cultured ELISpot assays were performed as previously described (5). In brief, peripheral blood mononuclear cells (PBMC) from healthy HLA-B*07+ donors (typed using low resolution polymerase chain reaction-based HLA-typing) were isolated through Ficoll, seeded at a density of 107 cells/mL in Dulbecco's modified eagle's medium (DMEM) (10% fetal calf serum), and stimulated with 1 μg/mL relevant peptide in individual wells. Interleukin-2 at a final concentration of 2 ng/mL (PeproTech, Hamburg, Germany) was added on day 2, 5, 7, 9, and 11. PBMC were harvested after 12 days and seeded at 5 × 105 cells/well in DMEM (10% fetal calf serum) onto nitrocellulose plates (Mabtech, Nacka Strand, Sweden). Stimulation was for 20 h with 1 μg/mL relevant peptide. Interferon (IFN)γ secretion was detected using an ELISpot kit (Mabtech), with spot numbers counted by an ImmunoSpot Series 6 ELISpot Reader (CTL, Bonn, Germany). PBMC that were positive by ELISpot assay were further expanded as previously described (19) using irradiated (100 Gy) K15 cells expressing HLA-C*0702, and preloaded with 1 μg/mL FRKDVNMVL peptide, for 2 weeks before staining with peptide-HLA multimers.
Peptide-HLA multimers, KIR multimers, and flow cytometry
HLA-C*0702 multimer constructs, encoding a C-terminal twin-strep-tag (IBA, Göttingen, Germany) and linked to the sequence of FRKDVNMVL in the vector pMXsIP were retrovirally transduced into J558L cells (5,18,19). Supernatants containing peptide-HLA multimers were concentrated ∼70-fold to 100 μL per staining using 100 kDa cutoff centricons (Merck Millipore, Darmstadt, Germany) and preincubated with 5 mL streptactin-phycoerythrin (IBA) before staining. Multimer staining of PBMC expanded on irradiated peptide-loaded K15 HLA-C*0702 feeders was performed in combination with CD3 antibody (BD Biosciences, Heidelberg, Germany), and cells in a living forward scatter/side scatter gate were analyzed on a Fluorescence-Activated Cell Sorter Calibur cytometer using CellQuest software (BD Biosciences). Since typing for HLA-C is not routinely performed by the Blood Bank at the University Hospital Tübingen, we made use of HLA-B*07+ HLA-A*02− donors, given that >90% of these donors are HLA-C*07+ (29), for these assays.
Alternatively, HLA-C*0702 streptamers containing the adenoviral peptide FRKDVNMVL or the control human peptide VRIGHLYIL from MAGE-A12 (36) were custom made by IBA. PBMC from healthy donors, typed for both HLA-B*07 and HLA-C*07, were expanded for 2 weeks using a peptide library of human adenovirus hexon protein (Miltenyi Biotech, Bergisch Gladbach, Germany). Cells were stained in combination with CD3 and CD8 antibodies and analyzed in a living forward scatter/side scatter gate using FlowJo software (FlowJo LLC, Ashland, OR).
The KIR2DL3 multimer construct (GeneCust) comprised amino acids M1-H245 fused via a linker IEGRMD to amino acids P100–P328 of human IgG1, mutated to prevent binding to Fc receptors and activation of C1q (L117A, L118E, G120A, P214S) (32), and a C-terminal Twin-strep-tag (IBA). KIR2DL3 multimer constructs in pMXsIP were retrovirally transduced into J558L cells, and supernatant containing KIR2DL3 multimer was concentrated ∼70-fold to 100 μL per staining using 100 kDa cutoff centricons (Merck-Millipore) and preincubated with 5 μL streptactin-phycoerythrin (IBA). Parental K15 cells or K15 HLA-C*0702 cells nonloaded or preloaded with FRKDVNMVL or FRKDVNMIL at a concentration of 10 μg/mL for 16 h were harvested and stained with KIR2DL3 multimers, and cells were analyzed in a living forward scatter/side scatter gate on a Fluorescence-Activated Cell Sorter Calibur cytometer using CellQuest software (BD Biosciences).
NK cytotoxicity assays
Parental K15 cells or K15 HLA-C*0702 cells nonloaded or preloaded with FRKDVNMVL or FRKDVNMIL at a concentration of 10 μg/mL were irradiated (100 Gy) and cocultured with NK-92MI KIR2DL3 cells at a ratio of 1:1 for 16 h. Samples were harvested and spiked with a defined number of polystyrene beads (diameter 9 μm; Polysciences, Eppelheim, Germany) to quantify cell numbers by flow cytometry as previously described (19). Cytotoxicity was assessed by determining the number of viable GFP+ target K15 cells in a forward scatter/side scatter living cell gate in the presence of NK cells relative that in the absence of NK cells.
Expression levels of HLA on cells infected with adenovirus
Hemagglutinin (HA)-tagged HLA-A*0201 and HLA-C*0702 constructs encoding the tapasin signal peptide and HA epitope sequences (MKSLSLLLAVALGLATAVSAGPAVYPYDVPDYAAALG) linked to the S26 residue in HLA were cloned into pMXsIP and retrovirally expressed in A549 lung epithelial cells. These cells were infected with human adenovirus 2 isolate BB2000-61 as previously described (19), and after 72 h, cells were harvested using phosphate-buffered saline containing 0.02% ethylenediaminetetraacetic acid. The expression of tagged HLA molecules was quantified by staining with fluorescein isothiocyanate (FITC)-coupled antibody to HA (BioLegend), compared to staining with FITC-coupled isotype control antibody X40 (BD Biosciences), using a fluorescence-activated cell sorter Calibur with gating on live cells in a forward scatter/side scatter gate.
Results
In our attempts to identify adenoviral peptide epitopes suitable for use in adoptive transfer therapy, we screened healthy HLA-B*07+ donors by cultured IFNγ ELISpot for responses to the peptide FRKDVNMVL from hexon protein (Fig. 1A, B). This peptide is found in species C and B adenoviruses, which are relevant in children and adults, respectively (23), and is conserved in other species with only one change in the penultimate residue. Three of six healthy donors clearly responded to this peptide (mean 518 ± SEM 111 spot forming cells) with responses comparable to that of the previously described HLA-B*0702-restricted peptide VPATGRTLVL (Fig. 1B) (19). These data confirm an earlier study showing T cell responses to FRKDVNMVL in healthy donors, and which suggested that the peptide is restricted by HLA-B*0702 (35). However, more recent prediction software suggested that the peptide may be restricted by HLA-C*0702 (NetMHCpan) (28), which shows strong linkage disequilibrium with HLA-B*0702 such that >90% of HLA-B*0702*+, HLA-A*0201− donors express HLA-C*0702 (29).
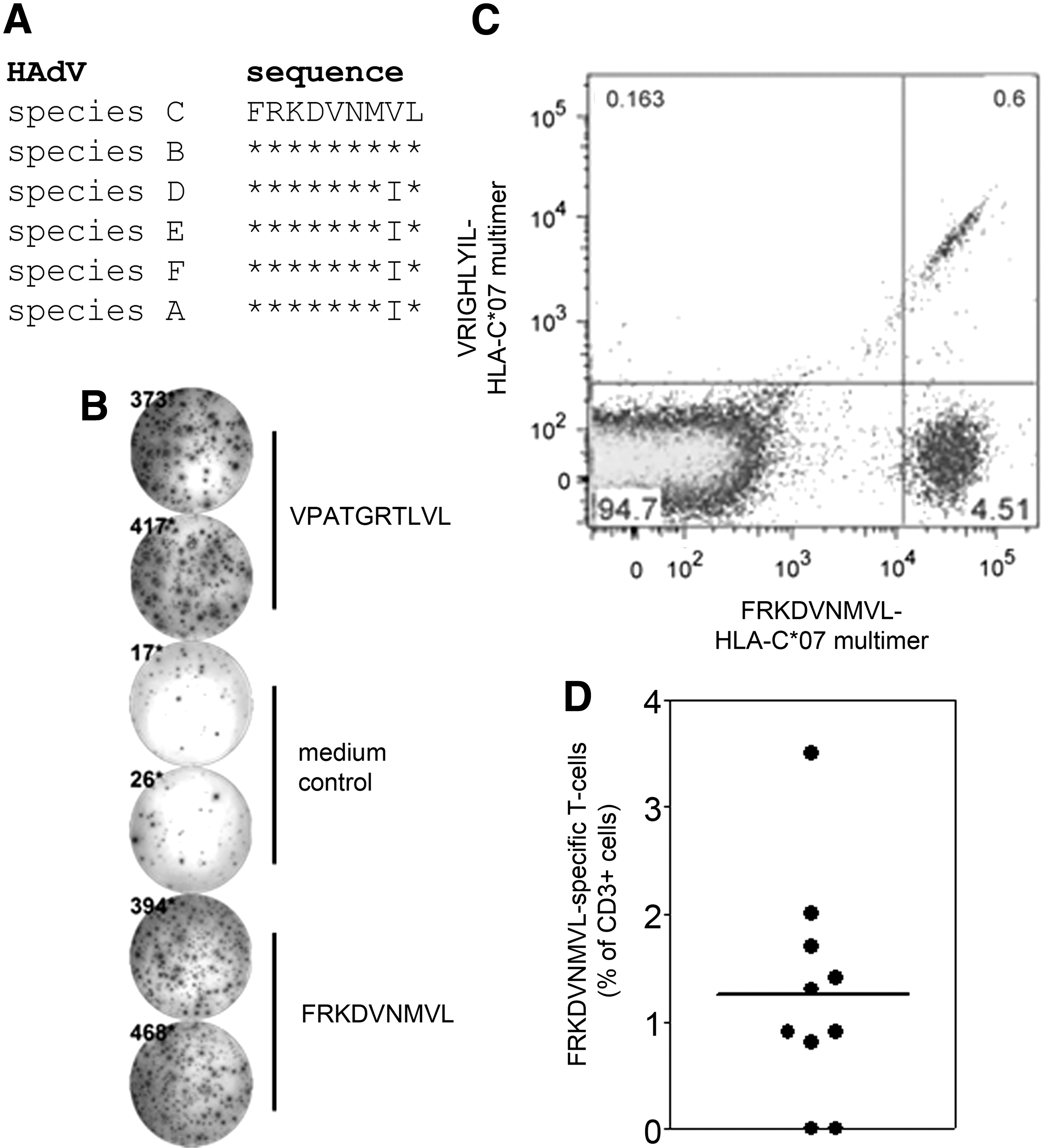
T cell responses to a conserved adenoviral epitope presented on HLA-C*0702.
To test whether FRKDVNMVL is restricted by HLA-C*0702, we generated peptide-HLA streptamers containing this and a negative control peptide VRIGHLYIL from the tumor antigen MAGE-A12 (36). Staining of PBMC from healthy HLA-B*07+ and HLA-C*07+ donors directly ex vivo did not detect defined T cell populations, suggesting that the frequency of FRKDVNMVL-specific T cells is low in healthy donors (data not shown). However, following expansion for 2 weeks with a peptide library of the adenovirus hexon protein, distinct populations of CD3+CD8+ T cells could be detected by FRKDVNMVL-HLA-C*0702 streptamer staining in two out of three donors (Fig. 1C).
A caveat of these assays is that HLA-C*0702 and its multimers can bind to KIR2DL3, which is expressed on both NK cells and highly differentiated T cells (4). We, therefore, also stained with a streptamer containing VRIGHLYIL from the tumor antigen MAGE-A12 to control for binding of the streptamers to KIR. A low percentage of CD3+ CD8+ cells stained positively with both streptamers, with no CD3+CD8+ cells positively stained with only the control streptamer containing VRIGHLYIL (Fig. 1C). These data suggest that the majority of CD3+CD8+ cells positively stained with FRKDVNMVL-HLA-C*0702 streptamers are peptide-specific T cells, and that a minority stained by both streptamers are differentiated T cells that express KIR2DL3.
As a further control, we stained KIR2DL2/3 on PBMC that responded to the FRKDVNMVL peptide. Only a minority (<10%) of CD3+ cells that stained positive with the FRKDVNMVL-HLA-C*0702 multimer expressed KIR2DL2/3 (data not shown), again suggesting that staining with FRKDVNMVL-HLA-C*0702 multimer detects T cells specific for the FRKDVNMVL peptide. To assess the prevalence of T cell responses, we expanded and stained specific T cells from multiple HLA-B*07+, HLA-A*02− donors, of which >90% express HLA-C*0702 (29). Although the frequencies of FRKDVNMVL-specific T cells were low in all donors, specific cells were detected in eight of ten donors indicating that such responses are immunodominant (Fig. 1D).
To confirm the restriction of FRKDVNMVL peptide by HLA-C*0702, we next determined whether this peptide can stabilize HLA on the cell surface. For this purpose, we generated HLA molecules covalently linked to defined adenoviral peptides as previously described (5,25) such that the effect of only one bound peptide can be assessed. The previously described peptide epitopes LTDLGQNLLY and VPATGRTLVL stabilized their restricting HLA allotypes (HLA-A*01 and HLA-B*0702, respectively) such that surface expression was comparable to that of the wild-type HLA molecules (Fig. 2). Surface expression of wild-type HLA-C*0702 was, however, lower than that of HLA-A*0101 and HLA-B*0702, but was increased ∼10-fold when linked to the peptide FRKDVNMVL (Fig. 2). A similar stabilization of HLA-C*0702 was seen when it was linked to the peptide FRKDVNMIL, which is found in adenovirus species other than species B and C (data not shown). Both of these peptides therefore stabilize HLA-C*0702 on the cell surface.

Stabilization of HLA-C*0702 by adenoviral peptides. K15 cells transduced with wild-type HLA-A*0101, -B*0702, or -C*0702 constructs (shaded histograms) or these constructs covalently linked to the peptides LTDLGQNLLY, VPATGRTLVL and FRKDVNMVL, respectively (unshaded histograms), were stained with pan-HLA class I antibody. Values from control staining of untransduced cells were less than MFI 13. MFI, median fluorescence intensity.
Given the surprising upregulation of HLA-C*0702 surface expression when covalently linked to adenoviral peptides, we wondered whether peptide-HLA-C*0702 complexes can modulate NK cell cytotoxicity. NK cells monitor the health of cells, in part, by sensing surface expression levels of HLA using killer inhibitory receptors—in this case KIR2DL3 (21).
We therefore tested whether the adenoviral peptides influence recognition of HLA-C*0702+ cells by staining with multimers of KIR2DL3. Preloading of cells with FRKDVNMVL or FRKDVNMIL peptides increased expression of HLA-C*0702 only marginally as determined by staining with pan-HLA class I antibody (Fig. 3B and data not shown). However, binding to KIR2DL3 multimers to cells preloaded with FRKDVNMVL was markedly enhanced compared with nonloaded cells (Fig. 3A). Such enhanced binding was also observed when cells were preloaded with FRKDVNMIL (median fluorescence intensity [MFI] 472 ± SEM 101 for FRKDVNMIL-loaded cells vs. MFI 445 ± SEM 87 for FRKDVNMVL-loaded cells vs. MFI 147 ± SEM 31 for nonloaded K15 HLA-C*0702 cells), and KIR2DL3 multimers did not bind to cells that were not transduced with HLA-C′0702 (Fig. 3A). Presentation of both adenoviral peptides on HLA-C*0702 molecules thus enhances binding to the inhibitory receptor KIR2DL3.

Presentation of the FRKDVNMVL peptide on HLA-C*0702 enhances binding to KIR2DL3 and decreases NK cytotoxicity.
To determine whether such enhanced KIR2DL3 binding affects NK activity, we performed cytotoxicity assays using KIR2DL3+ NK cells as effectors (Fig. 3C). As targets, we used K15 cells, variants of the HLA-negative K562 cell line that were either transduced with wild-type HLA-C*0702 or not. K15 cells were significantly protected from NK cytotoxicity if they expressed HLA-C*0702. However, preloading of HLA-C*0702 with FRKDVNMVL peptide significantly further decreased NK cytotoxicity. Preloading with FRKDVNMIL also resulted in apparently decreased NK cytotoxicity, although this was not as pronounced as that following preloading with FRKDVNMVL (Fig. 3C). Taken together, the enhanced binding to KIR2DL3 and concomitant decrease in NK cytotoxicity following presentation of FRKDVNMVL on HLA-C*0702 suggest that adenoviruses may use this peptide to evade the NK cytotoxic response.
Finally, given the stabilization of HLA-C*0702 by FRKDVNMVL or FRKDVNMIL peptides, we determined whether HLA-C*0702 expression levels at the cell surface are altered during infection with adenovirus. Since there is no HLA-C*07-specific antibody available, we used HA-tagged HLA constructs expressed on lung epithelial A549 cells. HLA-C*0702 expression levels on uninfected cells were again low compared with those of HLA-A*0201 and were not altered during adenovirus infection (Fig. 4, MFI 43 ± SEM 2 vs. MFI 45 ± SEM 7). By contrast, downregulation of HLA-A*0201 as a positive control was readily detected during adenoviral infection (Fig. 4, MFI 130 ± SEM 9 vs. MFI 50 ± SEM 9). These data suggest that adenovirus is not able to downregulate HLA-C*0702 expression to evade the T cell response during infection.
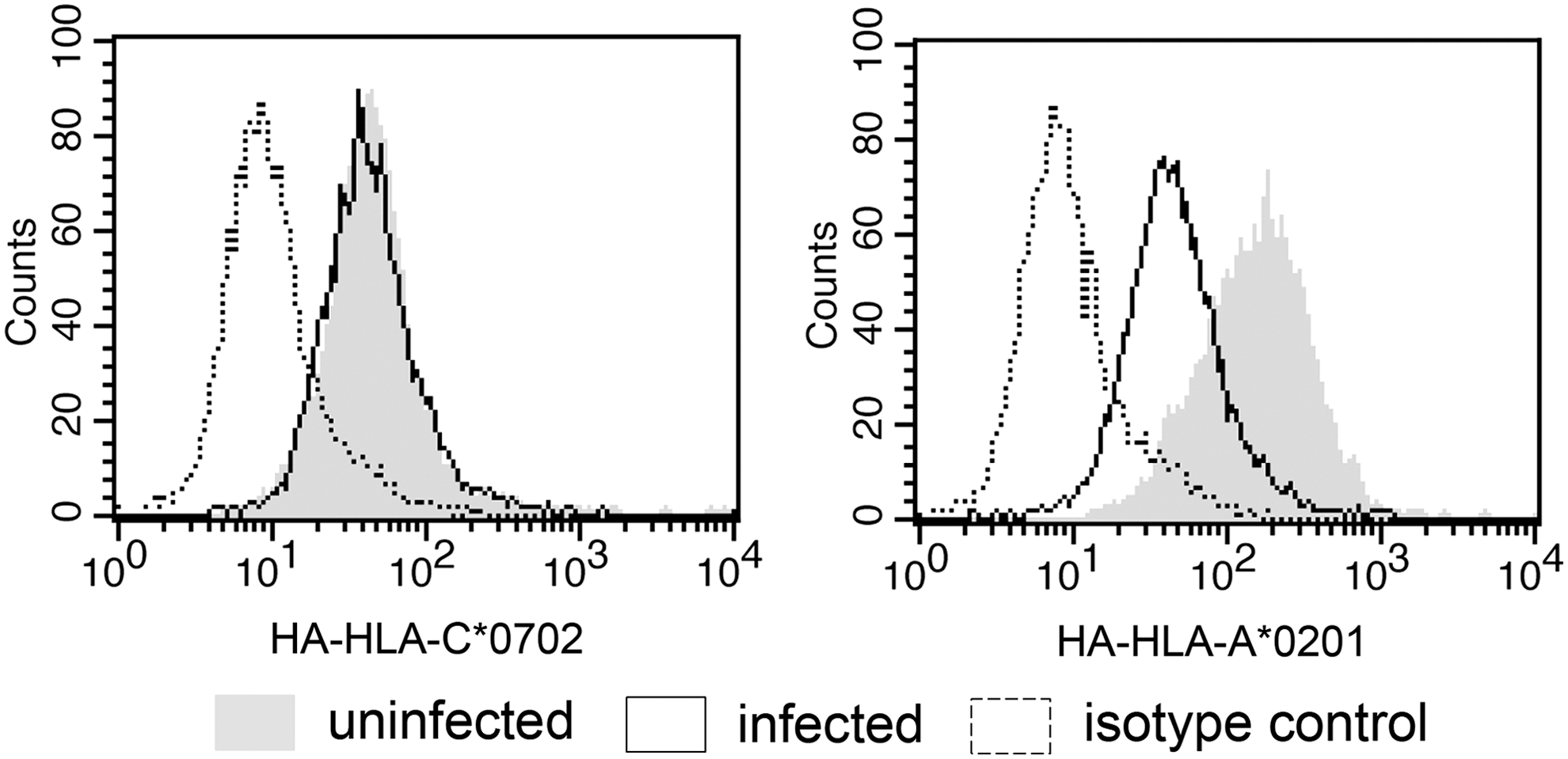
HLA-C*0702 is not downregulated on cells infected with adenovirus. A549 cells ectopically expressing HA-HLA-C*0702 (left) or HA-HLA-A*0201 (right) were either mock infected (shaded histograms) or infected with human adenovirus 2 (unshaded histograms with solid lines). After 72 h, cells were stained with an antibody to the HA tag. Unshaded histograms with dotted lines indicate isotype control stainings. Data from one of three independent experiments are shown.
Discussion
Adoptive transfer of specific T cells is a particularly effective treatment option for adenovirus infection in patients following hematopoietic stem-cell transplantation (13,14,26). However, there are still only a few well-defined peptide epitopes from adenovirus available for either immunomonitoring or direct selection of T cells for therapy, and arguably only one defined adenoviral epitope has been shown to induce protective T cell responses required for successful adoptive transfer therapy (19).
Many viruses, including adenoviruses evade T cell responses by downregulating HLA expression on infected cells (2,6). Coordinated surveillance of HLA-C allotypes by peptide-specific T cells and NK cells may counteract such immunoevasion, as described during HIV infection (21). We reasoned that HLA-C-restricted T cells may control infection better than T cells restricted by HLA allotypes not recognized by NK cells; therefore, we searched for adenoviral peptide epitopes restricted by HLA-C. We were particularly interested in the HLA-C*0702 allotype, which has the fourth highest allelic frequency in Europe (29).
We show that the peptide FRKDVNMVL from hexon protein of species B and C adenoviruses is presented by HLA-C*0702. Peptide-specific T cells could be detected in a majority of HLA-typed donors, indicating that such responses are immunodominant (Fig. 1C, D). Responses could also be detected by IFNγ ELISpot assays, suggesting that such specific T cells are functional (Fig. 1B). T cell responses to this peptide have previously been demonstrated and were suggested to be restricted by HLA-B*0702 (35). The reason why the restriction by HLA-C*0702 was missed, despite considerable effort to define the restricting HLA, is not clear. However, it is possible that some of the donors in that study, which were typed at low resolution, expressed not HLA-C*0702 but other allotypes such as HLA-C*0701, which have slightly different peptide binding motifs (28). A similar misallocation of the restriction of the human cytomegalovirus (HCMV) epitope CRVLCCYVL, also restricted by HLA-C*0702 and not by HLA-B*0702, was previously made for possibly the same reason (1).
Surface expression levels of HLA-C*0702 are low compared with that of HLA-A*0101 or HLA-B*0702, consistent with previous studies (3), but were enhanced ∼10-fold when FRKDVNMVL or its variant FRKDVNMIL was covalently linked to HLA-C*0702 (Fig. 2, data not shown). The reason for this enhancement is not clear, especially, since it was not seen with HLA-A*0101 or HLA-B*0702 covalently linked to well-described high-affinity peptide epitopes. These data suggest that the pool of endogenous peptides that can be presented on HLA-C*0702 may limit expression levels of this allotype on the cell surface.
Given the stabilization of HLA-C*0702 by both adenoviral peptides, we wondered whether presentation of such peptides on HLA-C*0702 affects NK cell responses. Although loading of the peptide FRKDVNMVL only marginally increased expression of HLA-C*0702, binding to multimers of KIR2DL3 was prominently enhanced (Fig. 3A). Consistent with this, NK cytotoxicity was significantly decreased in comparison to that induced by cells expressing nonloaded HLA-C*0702 alone (Fig. 3C). These data suggest that adenoviruses may use the presentation of the FRKDVNMVL peptide as a strategy to evade NK cell responses.
Although binding of KIR to HLA is not peptide specific, binding may nevertheless be influenced by residues in the C-terminal region of the peptide, with larger or charged residues inhibiting the interaction with KIR (8). The variant peptide FRKDVNMIL is expressed in species A, D, E, and F adenoviruses. Given the difference between the variant peptides at the penultimate residue, with the larger isoleucine instead of valine, we hypothesized that presentation of FRKDVNMVL would more efficiently inhibit NK responses than that following presentation of FRKDVNMIL. This was indeed the case (Fig. 3C). However, binding of KIR2DL3 multimers to HLA-C*0702 loaded with FRKDVNMVL or FRKDVNMIL was comparable. There is thus a discrepancy between the binding of KIR2DL3 multimers and inhibition of NK cytotoxicity by FRKDVNMIL-loaded cells, for which we have no obvious explanation.
However, we should point out that children are persistently infected with predominantly species C adenoviruses (23), which encode the FRKDVNMVL peptide that more efficiently inhibits NK responses. It remains possible that the FRKDVNMVL peptide is a factor that contributes to maintaining persistent infection in children by facilitating evasion of the NK response.
We finally wondered whether adenovirus is able to evade T cell responses to these peptides by downregulating HLA-C*0702 expression. Surprisingly, HLA-C*0702 surface expression levels were not downregulated during infection, in contrast to that of HLA-A*0201 as a positive control (Fig. 4). The use of HA-tagged constructs for this assay was necessary since there is no HLA-C*07-specific antibody available. However, while this assay is very specific for the relevant HLA allotypes, it is not very sensitive and may not have detected slight decreases in HLA-C*0702 expression, a problem which is compounded by its low expression levels. Nevertheless, these results are consistent with other reports showing that HLA-C*0702 is not functionally downregulated during HCMV infection, as measured by comparable inhibition of NK cytotoxicity before and after infection (1).
We note that reports of HLA-C downregulation during viral infection have been controversial. For example, it was generally accepted that HLA-C is not downregulated during HIV infection (3). However, more recently it has been shown that downregulation of HLA-A/B and HLA-C by HIV occurs through distinct mechanisms (2). While there may be distinct mechanisms that regulate HLA-C expression during adenoviral infection, we do not detect downregulation of HLA-C*0702 within the sensitivity of our assay system.
There are a number of ways, in which the FRKDVNMVL specificity could be used for adoptive transfer immunotherapy. First, given the high frequency of HLA-C*0702, the FRKDVNMVL specificity could be used to monitor the enrichment of specific T cells using the currently available protocols of expansion in vitro, or IFNγ capture technology before adoptive transfer (13,26). Such monitoring would provide 68% population coverage in Europe if combined with previously described hexon peptide epitopes (restricted by HLA-A*01, -A*24, -B*07, and–B*35) (16) thereby, making immunomonitoring at the level of single peptide epitope specificities possible.
Second, an attractive option for adoptive transfer therapy is selection of specific T cells using peptide-HLA streptamers, thus allowing donor lymphocyte infusion of nonstimulated specific T cells. Although the low frequency of adenovirus specific T cells makes such selection challenging, it has been successfully performed for one compassionate care treatment patient as well as in preclinical studies for at least some adenoviral peptide epitopes (7,16,19). Nevertheless, the low frequency of FRKDVNMVL-specific T cells that we observe in this study may well preclude such selection, although this remains to be demonstrated. Recently, the use of T-cell receptor transduced donor cells for adoptive transfer therapy of adenoviral infection has been explored, and we envisage that FRKDVNMVL may be a relevant target in this system (10).
Taken together, we describe an adenoviral peptide epitope FRKDVNMVL, which may facilitate evasion of the NK cytotoxic response by enhancing binding of its restricting HLA-C*0702 allotype to KIR2DL3. Furthermore, the immunodominance of FRKDVNMVL-specific T cell responses, apparent lack of HLA-C*0702 downregulation during infection, and the high frequency of the HLA-C*0702 allotype suggest that this peptide epitope may be particularly useful for adoptive T cell transfer therapy of adenovirus infection.
Footnotes
Acknowledgments
Supported by the German Center for Infection Research (DZIF) (K.M.D., D.H.B., M.N.). Use of human blood products was approved by the ethics commission of the Medical Faculty, University Hospital Tübingen. We thank the staff of the Blood Bank (Tübingen, Germany), Dario Campana (St. Jude Children's Research Hospital, Memphis, TN), Tim Greten (NIH, Bethesda, MD), Klaus Hamprecht (Tübingen, Germany), Elizabeth Jaffee (Johns Hopkins University, MD), Peter Lang (Tübingen, Germany), Josef Mautner (Munich, Germany), Andreas Moosmann (Munich, Germany), Wolfgang Schamel (Freiburg, Germany), and Annalena Wallisch (Tübingen, Germany).
Author Disclosure Statement
No competing financial interests exist.