
Abstract
The innate immune system is the first line of defense against virus infection that triggers the expression of type I interferon (IFN) and proinflammatory cytokines. Pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns, resulting in the induction of innate immune responses. Viral RNA in endosomes is recognized by Toll-like receptors, and cytoplasmic viral RNA is recognized by RIG-I-like receptors. The host innate immune response is critical for protection against virus infection. However, it has been postulated that an excessive inflammatory response in the lung caused by the innate immune response is harmful to the host and is a cause of lethality during influenza A virus infection. Although the deletion of genes encoding PRRs or proinflammatory cytokines does not improve the mortality of mice infected with influenza A virus, a partial block of the innate immune response is successful in decreasing the mortality rate of mice without a loss of protection against virus infection. In addition, morbidity and mortality rates are influenced by other factors. For example, secondary bacterial infection increases the mortality rate in patients with influenza A virus and in animal models of the disease, and environmental factors, such as cigarette smoke and fine particles, also affect the innate immune response. In this review, we summarize recent findings related to the role of PRRs in innate immune response during respiratory viral infection.
Introduction
D
In the cytoplasm, viral RNAs are sensed by RLRs. RLRs comprise retinoic acid-inducible gene-I (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). RIG-I and MDA5, which are cytoplasmic RNA helicases, bind viral double-stranded RNA (dsRNA) and triggers the signal to induce the innate immune response (177,178). dsRNA molecules generated by influenza A virus, VSV, respiratory syncytial virus (RSV), Newcastle disease virus (NDV), Japanese encephalitis virus, and HCV are sensed by RIG-I, whereas those of dengue virus and WNV are recognized by both RIG-I and MDA5, which signal through the mitochondrial antiviral-signaling protein (MAVS) adaptor molecule, resulting in the production of type I IFN (64,87).
The activation of these viral RNA sensors is strictly regulated, and autoimmune disorders are known to occur as a result of their aberrant activation (33,66). Moreover, these PRRs promote early inflammation in the lungs during viral infection (76). Dendritic cells (DCs) and macrophages expressing PRRs are important in the early inflammatory process (76). Although TLRs exhibit cell type–specific expression pattern, RLRs are widely expressed in various types of cells, such as DCs, macrophages, epithelial cells, and fibroblasts (63,68,88). The innate immune response is essential for controlling viral infection; however, excessive inflammation caused by a strong innate immune response is harmful to the host and is a cause of lethality in influenza A virus-infected patients (62,70). An accumulating body of evidence elucidates the underlying mechanisms of control of viral replication and virus-mediated excessive inflammation by the innate immune response. In this review, we summarize these recent findings and discuss the underlying mechanism of the excessive innate immune response.
RIG-I-Like Receptors
The RIG-I and MDA5 proteins contain caspase activation and recruitment domains (CARDs), which are essential for their association with the adaptor molecule, MAVS, resulting in the activation of downstream signaling (67,88,97,143,173). On the other hand, LGP2 does not contain CARDs, and thus plays regulatory roles during the activation of RIG-I and MDA5 (10,136,164). In addition, RIG-I, MDA5, and LGP2 contain a helicase domain and a C-terminal domain (CTD) that binds to viral RNA (18,152,178).
During replication of viral genomic RNA, dsRNA is generated by the viral RNA-dependent RNA polymerase; in addition, panhandle structure in the single-stranded genomic RNA of several types of viruses is reported to make a short dsRNA region (137,167). These cytoplasmic viral dsRNAs are recognized by RLRs (88,177,178). The RIG-I protein binds to relatively short dsRNAs (<1 kbp), whereas MDA5 binds to longer dsRNAs (>1 kbp) (Fig. 1) (65), suggesting that the difference in ligand specificity is one reason for the differential role of RIG-I and MDA5 in the recognition of virus RNA.

Pattern recognition receptors for viral RNA. TLR3, 7, and 8 are localized in endosomes. Endosomal dsRNA is recognized by TLR3, whereas ssRNA is recognized by TLR7 and TLR8. TLR3 requires TICAM-1/TRIF adapter protein to induce downstream signaling, meanwhile, TLR7 and 8 utilizes MyD88 adaptor for signaling. Cytoplasmic viral dsRNA is recognized by RIG-I and MDA5. dsRNA molecules shorter than 1 kbp are preferentially recognized by RIG-I, whereas dsRNA molecules longer than 1 kbp are recognized by MDA5. Both RIG-I and MDA5 use MAVS adaptor protein to trigger downstream signaling. dsRNA, double-stranded RNA; ssRNA, single-stranded RNA; RIG-I, retinoic acid-inducible gene-I; MAVS, mitochondrial antiviral-signaling protein.
Various types of viruses induce the stress granule formation after viral infection (104). Onomoto et al. have shown that RIG-I and MDA5 are recruited to stress granules after viral infection and sense viral RNA within stress granules (103). Therefore, they termed it antiviral stress granules (avSG). In addition to avSG, viral replication complexes (vRC), which contain both positive- and negative-stranded viral RNAs, are also targeted by RIG-I (100). vRC form a few hours after viral infection and is followed by avSG formation. Thus, it was postulated that RIG-I senses viral RNA within vRC and subsequently senses viral RNA within avSG (100).
After recognition of viral RNA, RIG-I and MDA5 assemble along the viral dsRNA molecule generating a nucleoprotein filament (116,171). The assembly of the proteins results in the formation of a 2CARD tetramer structure consisting of four RIG-I or MDA5 molecules (116,171). The RIG-I 2CARD tetramer structure is stabilized by a TRIM25-mediated K63-linked polyubiquitin chain (Fig. 2) (117). TRIM25 is an E3 ubiquitin ligase essential for RIG-I-mediated type I IFN production (34). Another ubiquitin ligase, Riplet, is also required for RIG-I activation (107,108). Riplet mediates K63-linked polyubiquitination of RIG-I at its C-terminal region, which promotes the binding of TRIM25 to RIG-I (111). MEX3C ubiquitin ligase also mediates K63-linked polyubiquitination of RIG-I at its N-terminal region (78). Although MEX3C is essential for RIG-I activation, the underlying mechanisms of this activation have not been fully elucidated (78). Assembly of MDA5 along viral dsRNA is regulated by phosphorylation. In resting cells, MDA5 is phosphorylated to suppress abnormal activation of MDA5 (169). Dephosphorylation of MDA5 by protein phosphatase 1 (PP1) is induced after viral infection, resulting in the activation of downstream signaling (169). Conversely, RIOK3, a protein kinase, mediates the C-terminal phosphorylation of MDA5, resulting in the disassembly of MDA5 and the downregulation of MDA5-mediated innate immune response (155). The mitochondrial targeting chaperone, 14-3-3ɛ, plays a crucial role in the translocation of RIG-I into the mitochondria (85). RIG-I and MDA5 require MAVS adaptor molecule, which is located on the outer membrane of the mitochondria, for the induction of the innate immune response (143). The 2CARD tetramer structure acts as a core for the prion-like aggregation of MAVS on the outer membrane of the mitochondria, which leads to the production of type I IFN and proinflammatory cytokines (Fig. 2) (50,117). Several cytoplasmic RNA helicases are involved in RIG-I-mediated type I IFN production (23,113), and the RIG-I pathway is linked to viral RNA degradation pathways (92,112,114).
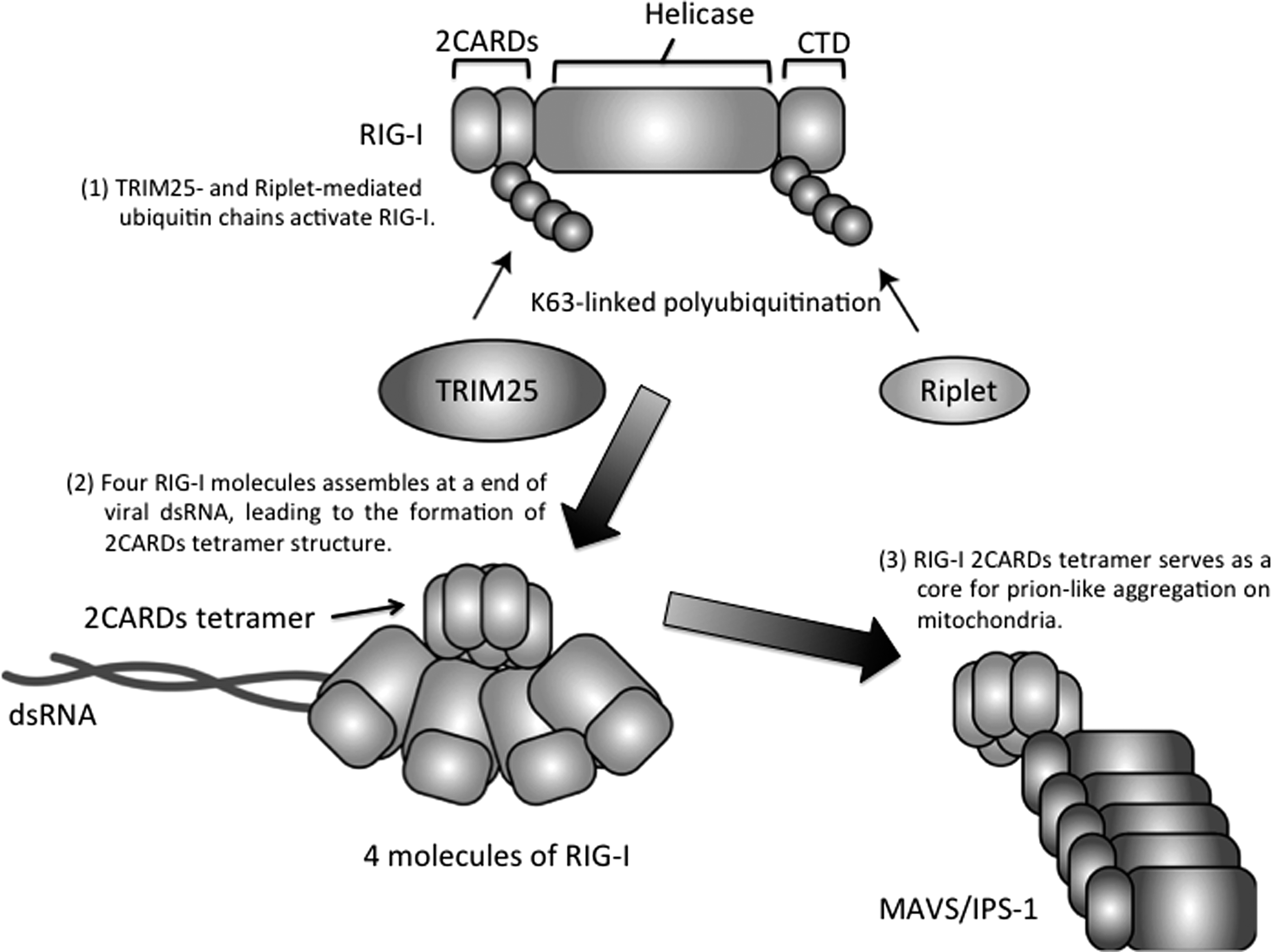
Molecular mechanism of RIG-I activation. The RIG-I protein comprises N-terminal two CARDs (2CARDs), a central helicase, and a CTD. The CTD recognizes viral dsRNA ends. RIG-I assembles along viral dsRNA, and the assembly of four RIG-I molecules results in the formation of a 2CARDs tetramer structure. The 2CARD tetramer structure acts as a core for prion-like aggregation of MAVS/IPS-1 on the surface of the outer membrane of mitochondria. Riplet ubiquitin ligase mediates K63-linked polyubiquitination of RIG-I at its C-terminal region, and TRIM25 ubiquitin ligase delivers a K63-linked polyubiquitin chain to the RIG-I-N-terminal region, which stabilizes the structure of 2CARDs tetramer. CTD, C-terminal domain; CARD, caspase activation and recruitment domain.
The LGP2 protein does not contain CARDs in its N-terminal region (178), hence it plays only a regulatory role. Ectopic expression of LGP2 suppresses RIG-I and MDA5 signaling (178). There are contradictory articles reporting the phenotype of LGP2 knockout mice (136,164). Venkataraman et al. first reported that knockout of LGP2 increased RIG-I-mediated signaling, and this is consistent to the study of LGP2 overexpression (164). Later, Satoh et al. reported that LGP2 knockout attenuated RIG-I and MDA5 signaling (136). Bruns et al. postulated a mechanism that LGP2 regulates MDA5 filament assembly, thereby promoting MDA5-mediated signaling.
Toll-Like Receptors
TLRs are type I transmembrane proteins, and their extracellular region comprises leucine-rich repeats and exhibits horseshoe structure (14,68). Cytoplasmic region of TLRs contain a TIR-domain, which is essential for binding to adaptor molecules and for the triggering of signals that induce an innate immune response.
Many viruses infect host cells through endocytosis. Hence, while RLRs recognize cytoplasmic viral dsRNA, TLRs recognize endosomal RNA. dsRNA and single-stranded RNA (ssRNA) are recognized by TLR3 and TLR7, respectively (Fig. 1). TICAM-1/TRIF is a solo adaptor of TLR3 capable of inducing IRF-3-mediated type I IFN production (105,175). In animal models, the TLR3–TICAM-1 signaling axis plays a crucial role in the antiviral innate immune responses against a picornavirus infection (1,110). However, the role of TLR3 in innate immune response to other viruses remains controversial. For instance, there are several contradictory reports about the role of TLR3 during WNV (19,166). In addition, impairment of TLR3 pathway in mice did not affect the innate immune response against influenza A virus infection (74).
Although TLR3 fails to bind ssRNA (3), TLR3 can sense viral ssRNA in some situations; this is because some types of viral ssRNA form short dsRNA regions, which are sufficient for recognition by TLR3 (157). In contrast to TLR3, TLR7 binds to ssRNA, and its expression is detected in plasmacytoid dendritic cells (pDCs) (25). TLR7 requires the MyD88 adaptor protein to trigger downstream signaling cascades (49). TLR8 is related to TLR7 and it recognizes viral ssRNA as TLR7 does, but in mice, TLR8 is not functional (47). TLR9 is also localized to endosomes and it recognizes nonmethylated CpG DNA and triggers the innate immune response through MyD88 adaptor protein (48).
TLR4 is a PRR for lipopolysaccharide of gram-negative bacteria; however, it also binds to viral proteins and to damage-associated molecular patterns, such as heat shock proteins, which are released from host cells damaged by virus infection (Fig. 1) (68,95). Unlike other TLRs, TLR4 utilizes four adaptor proteins, TICAM-1/TRIF, TICAM-2/TRAM, MyD88, and Mal/TIRAP (31,106,174 –176). TICAM-1/TRIF itself cannot bind to the TIR domain in TLR4, but to TICAM-2/TRAM that bridges TICAM-1/TRIF and TLR4 (106). Thus, TICAM-1 and TICAM-2 adaptors are important for TLR4-mediated type I IFN production.
Other PRRs Recognizing Viral RNA and DNA
TLRs and RLRs play crucial roles for the recognition of various types of viruses (68,88), whereas several other sensor molecules have also been reported. DEAD-box helicase 3 (DDX3) is a cytoplasmic RNA helicase. First it was reported that DDX3 is involved in RIG-I-mediated innate immune responses (140,148). Later, it was shown that DDX3 bound to viral RNA and promoted RIG-I activation (109). Recently, Gringhuis et al. showed that DDX3 binds to viral RNA of a lentivirus, thereby activating the MAVS adaptor molecule (44), suggesting that DDX3 is a sensor for viral RNA. It is also reported that NOD2, a member of NOD-like receptors, also sense viral RNA and trigger the innate immune response (131).
Cytoplasmic viral DNA is mainly recognized by cyclic-GMP-AMP synthase (cGAS) (83,139). The cGAS protein binds to viral DNA and produces a second messenger, cyclic-GMP-AMP (cGAMP) (150,172). The second messenger associates with stimulator of interferon genes (STING) on endoplasmic reticulum, which results in type I IFN production (150). Absent in melanoma 2 (AIM2) is another DNA sensor. Unlike cGAS, AIM2 drives the assembly of multiprotein complexes called inflammasome, which results in the activation of caspase-1 required for mature IL-1β production (11). AIM2 senses not only viral DNA but also host DNA damage caused by radiation (51).
Knockout of cGAS markedly reduced type I IFN production during DNA virus infection (83). However, there are several reports showing the involvement of other sensor molecules in the recognition of viral DNA. DNA-PKcs and Ku70/80 proteins bind cytoplasmic viral DNA and induce type I and type III IFN production, respectively (30,179). Interferon-inducible gene 16 (IFI16) is a member of AIM2 family. Unterholzner et al. first reported that IFI16 is a cytoplasmic viral DNA sensor (162). Later studies reported crucial roles of IFI16 in the innate immune response against several types of viruses (17, 37, 54, 147). However, Gray et al. recently questioned the role of IFI16 in antiviral innate immune response. They reported that deletion of mouse AIM2-like receptor locus, which includes AIM2 and IFI16 genes, reduced IL-1β production in response to cytoplasmic DNA, but failed to reduce IFN-β mRNA expression of mouse bone marrow-derived macrophages in response to cytoplasmic DNA (42). They also showed that knockout of human IFI16 gene in human primary fibroblasts failed to reduce IFN-β mRNA expression in response to cytoplasmic DNA (42). However, another group reported that knockout of human IFI16 in THP-1 cells markedly reduced IFN-β mRNA expression in response to cytoplasmic DNA. Moreover, Almine et al. reported that IFI16 in keratinocytes played a crucial role for type I IFN production in response to cytoplasmic DNA (4). These data imply the cell type–specific role of IFI16 in the innate immune response.
Virus-Mediated Inhibition of Host Innate Immune Response
TLRs and RLRs recognize viral infections and are essential for controlling them. On the other hand, viruses have evolved to escape the host innate immune response. For instance, NS3-4A of HCV cleaves MAVS and TICAM-1/Trif to escape host innate immune responses (27,28,82,97,133), and dengue virus suppresses 14-3-3ɛ-mediated RIG-I translocation into the mitochondria, resulting in the downregulation of type I IFN expression (12). These viral abilities to escape host innate immune responses have also been observed in respiratory viruses, such as influenza A virus. The NS1 protein of influenza A virus suppresses RIG-I signaling (24) by inhibiting TRIM25 and Riplet-mediated K63-linked polyubiquitination of RIG-I (35,123). MV is transmitted through the respiratory rout. The viral V protein of MV inhibits PP1-mediated MDA5 dephosphorylation, resulting in the attenuation of MDA5 signaling (20,169). Although these mechanisms benefit the virus in the infected host cells, PRRs in DCs and macrophages can still induce innate immune response as described below.
PRRs Expressed in DCs and Macrophages
DCs can be categorized as plasmacytoid, conventional, or monocyte-derived DC (pDC, cDC, or moDC, respectively) (7,72). pDCs rapidly produce large quantities of type I IFNs in response to viral infection (146). Human pDCs selectively express TLR7 and TLR9, but not other TLRs, and murine pDCs also express TLR7 and TLR9 (39). cDCs are categorized into several subsets as described below. In these different DC subsets, TLR3 exhibits cell type–specific expression. TLR3 is expressed in mouse splenic CD8α + DCs and human BDCA3 (CD141)+ DCs in blood (58,141). Two subsets of cDCs, CD103+ DCs and CD11b+ DCs, are present in the lungs (72). Lung CD103+ DCs express TLR3; on the contrary, CD11b+ DCs barely express TLR3, but express TLR7 instead (Fig. 3) (22). Circulating monocytes in blood stream can give rise to lung moDCs in a steady state (163). Because moDC also expresses CD11b, it is easily contaminated with CD11b+ cDCs (126). TLR3, TLR8, and TLR9 are expressed in moDCs, which also express RLRs (16,21,57) and have the ability to produce type I IFN in response to infection with influenza A virus or Sendai virus (15,16).
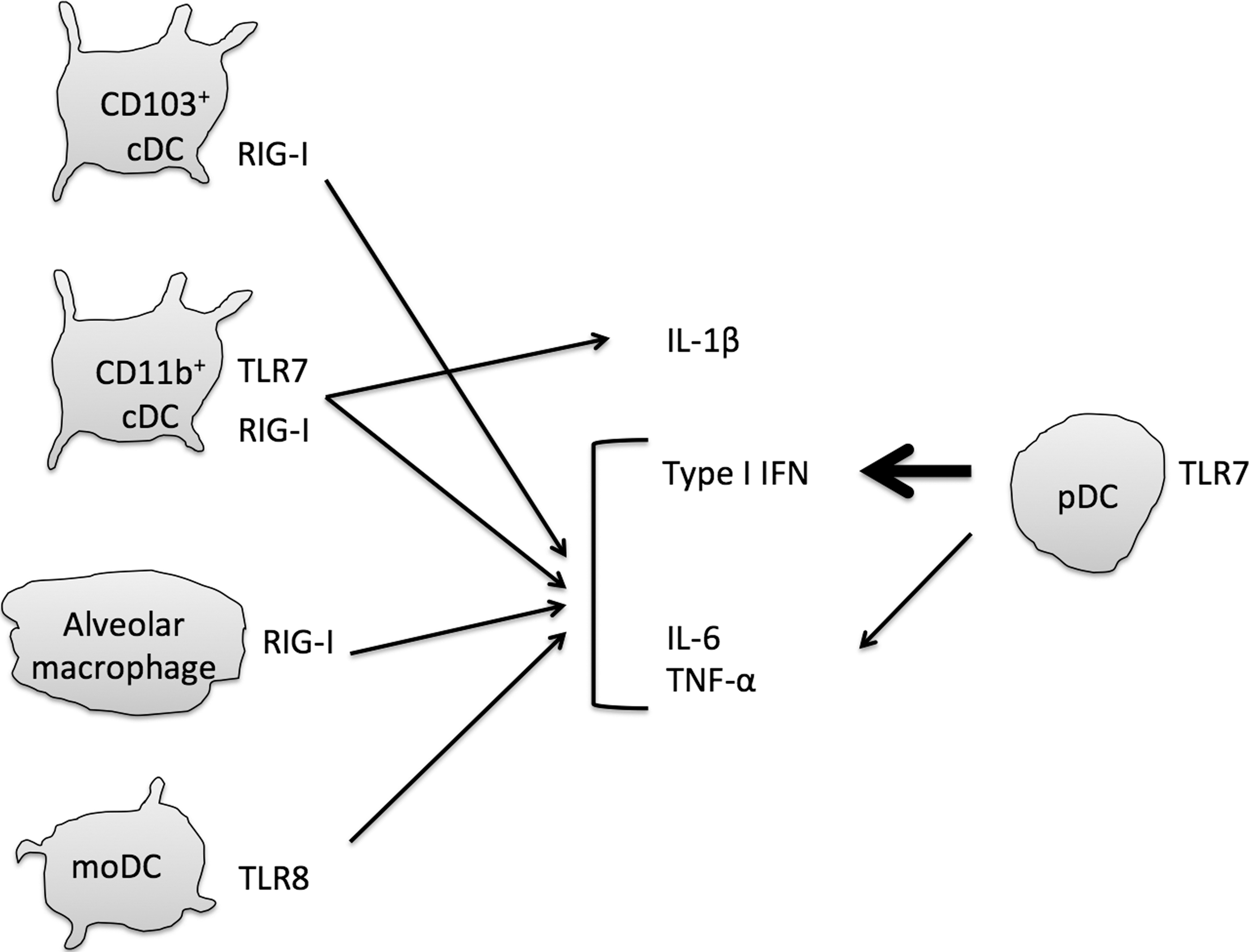
Interferon and cytokines produced by macrophages and dendritic cells during influenza virus infection. Influenza A virus is recognized by RIG-I, TLR7, and TLR8. RIG-I is ubiquitously expressed in various types of cells. Alveolar macrophages require RIG-I to produce type I IFN, IL-6, and TNF-α in early infection. CD103+ cDCs also require RIG-I. In contrast, moDCs require TLR8 to produce type I IFN and proinflammatory cytokines. CD11b+ cDCs use TLR7 and RIG-I to produce IL-1β as well as type I IFN and proinflammatory cytokines. pDCs produce large amounts of type I IFN and require TLR7 to sense influenza virus infection. cDC, dendritic cell conventional; pDC, plasmacytoid dendritic cell.
TLRs and RLRs are important for DC maturation. CD80 and CD86 are costimulatory molecules required for priming of naive T cells. Virus infection increased the expression of costimulatory molecules in DCs, and malfunction of RLRs attenuates the expression of costimulatory molecules (77). TLR7 is also important for naive T cell activation (74). TLR3 plays crucial roles in cross priming of naive CD8+ T cells that result in the activation of cytotoxic T cells against virus-infected cells (141). Therefore, CD103+ DCs are important for the CD8+ T cell-mediated antiviral response (72). TLR3 expressed in cDCs is also required for NK cell activation through the INAM molecule (29,61). Function of moDC in viral infection in the lung has not been fully elucidated; however, it has been shown that moDCs in the lung play a crucial role for initiating Th2 response to house dust (119).
In the lung, there are three types of macrophages, alveolar macrophages, bronchial macrophages, and interstitial macrophages (6). Bronchial macrophages are isolated from induced sputum, and interstitial macrophages are localized in the interstitium (32). Alveolar macrophages are found in alveolar lumen and play a role in innate immune responses (6). In general, macrophages are classified into M1 and M2 macrophages (93). M1 macrophages produce type I IFN and proinflammatory cytokine, whereas M2 macrophages are able to produce an immunosuppressive cytokine, IL-10 (93). Alveolar macrophages are also classified into M1 and M2 macrophages (6). Previous studies have revealed an important role of M1 alveolar macrophages during respiratory virus infection (76,165). M1 alveolar macrophages express RLRs and produce type I IFNs (Fig. 3) (76). Kumagai et al. have shown that murine alveolar macrophages act as a type I IFN producer important for the initial responses to viral infection in the lung, and that pDCs function when alveolar macrophage-mediated type I IFN production is abrogated (76). RLRs are expressed not only in macrophages but also in other types of cells.
Innate Immune Response to Respiratory Viruses
RLRs and TLRs are involved in the recognition of RSV infection. Sasai et al. first reported that both RIG-I and MDA5 were involved in RSV-mediated type I IFN expression; however, later studies reported that RIG-I plays a major role in the recognition of RSV infection compared with MDA5 (86,87,135). Several studies have implied the involvement of TLR7 for sensing RSV RNA (90,138). Knockout of RIG-I adaptor MAVS abolishes IFN-β production from lung fibroblasts, bone marrow-derived macrophages, and cDCs after RSV infection (8). Moreover, IFN-β and IL-6 expression in the lung of RSV-infected mice in vivo were markedly reduced by MAVS knockout. In contrast, MyD88 is dispensable for lung IFN-β and IL-6 expression after RSV infection (8). However, both MAVS and MyD88 are required for the IL-1β and TNF-α expression in the lung, and viral titers were increased by MAVS and MyD88 double KO compared with each single KO (8). It is intriguing that antibodies and CTL against RSV were reduced, but were still detected even in the DKO mice (8). These observations suggest that TLR3 and/or other PRRs are also involved in innate immune responses, including upregulation of costimulatory molecules and antigen presentation by DCs.
Human metapneumovirus (HMPV) is a respiratory virus and belongs to the family of Paramyxoviridae. RIG-I recognizes HMPV and induces type I IFN expression through IRF and NF-κB transcription factors (41,84), and MDA5 is dispensable for HMPV-induced type I IFN expression (41). Although viral replication is dispensable for RIG-I recognition of viral RNA in several types of viruses, replication of HMPV is essential to be recognized by RIG-I (41). TLR7 in pDCs also plays a crucial role of type I IFN production in response to HMPV (41).
Mice are not susceptible for MV infection, and thus several groups sought to establish mouse models by using knockout or transgenic mice. Several measles virus strains infect host cells through human CD46 or CD150 membrane proteins, and Seya and colleagues reported that human CD46 and CD150 transgenic (Tg) mice were sensitive to MV once MV-infected DCs were injected to the Tg mice (153, 154). This is consistent to the observations that DCs and alveolar macrophages are primary targets of MVs in nonhuman primate animal models. It is postulated that MV-infected DCs migrated secondary lymphoid tissues, such as bronchial alveolar lymphoid tissue and draining lymph node, where MV infects and replicates in B and T cells (80). Although human CD150 Tg mice are slightly permissive to MV, knockout of the gene encoding IFN-α receptor (IFNAR) in human CD150 Tg mouse markedly increases the susceptibility to MV, indicating that type I IFN is responsible for the protection against MV infection in the mouse model (168). Takaki et al. generated several knockout mice in human CD150 Tg background mice, and found that CD4+ DCs isolated from mouse spleen could sense MV infection and produced type I IFN through a MyD88-dependent pathway and that TLR3- and RLR-dependent pathways played only a minor role for the protection (154). These observations indicate that the TLR7–MyD88 pathway in DCs and/or macrophages is important for antiviral innate immune response to MV.
DC-SIGN is a C-type lectin and sense MV, which results in the activation of Raf1, a serine–threonine kinase (43). Raf-1 inhibits GAD33-PP1 holoenzyme activation, which is required for RLRs phosphorylation (96). Hence, MV efficiently infects human DCs by inhibiting RLR-induced type I IFN production by DC-SIGN activation (96). The V protein of MV also has abilities to suppress RLR-mediated type I IFN production (20,153). These observations are consistent with the notion that TLR7–MyD88 pathway, but not RLR pathway, is essential for type I IFN production during MV infection.
RIG-I is expressed in fibroblasts, macrophages, and DCs, and recognition of influenza A virus RNA by RIG-I results in the production of type I IFN and IL-6 (63,64,108). Studies using knockout mice indicated that the TLR3-TICAM-1 pathway in cDCs plays only a minor role in the antiviral innate immune response against influenza A virus infection (74), and cDCs require RIG-I to sense influenza A virus RNA. In contrast, pDCs require TLR7 to recognize influenza A virus, resulting in the type I IFN production (Fig. 3) (25). In fact, pDCs are able to produce large amounts of type I IFN in response to influenza A virus infection, and knockout of TLR7 and MyD88 genes leads to a substantial reduction in type I IFN serum levels during influenza A virus infection in vivo (74). In the lung tissue of mice infected with influenza A virus, both RIG-I- and TLR7-mediated signaling pathways contribute to the production of type I IFN and to the expression of IFN-inducible genes. However, TLR7-, but not RIG-I-, signaling is responsible for the production of antibodies against the HA of influenza A virus in serum (74). In addition, TLR7, but not RIG-I, plays a crucial role in the activation of NP-specific CD4+ T cells. However, TLR7 and RIG-I play only a minor role in the activation of NP-specific CD8+ T cells (74).
Influenza virus infection induces the production of proinflammatory cytokines, such as IL-1β. TLR7 is required for the synthesis of pro-IL-1β (53), which is processed by caspase-1 to release the mature IL-1β. The M2 protein of influenza A virus perturbs the ionic concentration in infected cells, resulting in the activation of the NLRP3 inflammasome, which then triggers caspase-1 activation (53). Production of IL-1β is essential for the activation of influenza A virus-specific CD8+ T cells. IL-1β stimulation induces migration of cDCs to lymph nodes, where they prime naive CD8+ T cells (115).
Influenza B virus is also important respiratory pathogens. As influenza A virus, influenza B virus is also recognized by RIG-I in mouse embryonic fibroblasts and HEK293T cells (91). Influenza B virus induces type I IFN production by human macrophages and moDCs (56,91). Knockout of TLR3 does not affect the production of specific antibodies nor the CD8+ T cell activation (46). Although TLR7 knockout reduced the specific antibody production, cytotoxic T cell activation is normal even in TLR7 mice (46). Therefore, TLRs and RLRs might play redundant roles in the activation of T cells or other PRRs are required for priming of naive T cells during influenza B virus infection.
Influenza A Virus-Mediated Excessive Innate Immune Responses
It has been shown that mice and macaques infected with the reconstructed 1918 influenza A virus show marked expression of proinflammatory cytokines, type I IFN, and IFN-inducible genes within a few days after infection (62,70). Enhanced expression of proinflammatory cytokines in response to H5 N1 pandemic virus has also been reported (36). Excessive production of cytokines caused by infection with influenza A virus, also called cytokine storm, has been widely hypothesized to be the main cause of pathology and, ultimately, of death (36,134).
Salomon et al. have investigated the effect of knocking out of TNF-α, TNF-R1, IL-6, and CCL2 genes on the survival of mice infected with H5 N1 (134). Even though these are key proinflammatory cytokines induced during H5 N1 infection, the knockout of any of these genes failed to improve the survival of H5 N1 infected mice (134). Treatment with glucocorticoids, which are cytokine inhibitors, also had no effect on the survival of infected mice (134).
On the other hand, it was reported that LGP2 overexpression improved the survival of mice infected with an H3/N2 influenza A virus strain (A/Scotland/20/74) (145). Si-Tahar et al. reported that LGP2 transgenic mice are more resistant to influenza A virus infection than wild-type mice, suggesting that attenuated RIG-I signaling rendered the mice resistant to influenza A virus infection (145). Although LGP2 is an RLR, it is also required for controlling the survival of CD8+ T cells during T cell expansion (151). Therefore, the underlying mechanism of LGP2-mediated resistance to influenza A virus is not fully elucidated.
In contrast to LGP2 transgenic mice, deletion of RLRs and TLRs does not improve the survival of mice. RIG-I knockout mice showed a marked reduction in the expression of IFN-β, CCL2, and IL-1β in the lung, and were more susceptible to infection with a sublethal dose of influenza A virus (PR8 strain) than wild-type mice (60). Mx1 transgenic mice are considered a more accurate model of influenza pathogenesis in humans than wild-type mice (118). This is because wild-type C57BL/6 mice carry nonfunctional allele of Mx1 gene, although Mx1 plays a crucial role for antiviral innate immune response in human cells (118). Knockout of both TLR7 and MAVS genes in Mx1 transgenic mice markedly reduced their survival after infection with influenza A virus (PR8) (118). In addition, the knockout of both TLR3 and TLR7 genes have been shown to reduce the survival of mice infected with influenza A virus PR8 strain (142), whereas the survival of MAVS knockout mice was unaffected (142).
In contrast to total knockout of PRRs, appropriate regulation of the innate immune response renders mice resistant to influenza A virus infection. Sphingosine-1 phosphate (S1P) receptors are a family of five G protein-coupled receptors targeted by S1P (129), and stimulation of S1p1 receptor decreases the cytokine expression and proliferation (128). S1P receptor is expressed on lymphocytes and endothelial cells, but not on DCs and macrophages in the lung (158). Administration of agonists of S1P receptor moderately reduces cytokine levels in bronchoalveolar lavage fluids (BALF) of mice infected with influenza A virus; however, it markedly increased the survival (158). A recent study showed that agonists for S1P receptor reduced the cytokine production in a TLR- and RLR-independent manner (159). These observations imply that endothelial cells and lymphocytes in the lung are responsible for lethal cytokine storm and that DCs and macrophages expressing PRRs play a protective role during influenza A virus infection (158). CARD9 is an adaptor of C-type lectin and is also involved in RIG-I signaling (130). CARD9 is required for NF-κB activation, but is dispensable for the activation of IRF transcription factors that are essential for type I IFN production (120,130). The deletion of CARD9 substantially reduced IL-6, TNF-α, CXCL1, and CXCL10, but not type I IFN production in BALF collected from the influenza A virus-infected mouse lungs, which improves survival of the infected mice (161).
Other Factors Involved in the Excessive Inflammation During Influenza A Virus Infection
Aside from influenza A virus infection itself, other factors can also affect the innate immune response to virus infection in the lung. Secondary bacterial infections increases the morbidity and mortality rates during influenza A virus infection (45,94). In case of novel flu infection in 2009, 20–50% of hospitalized virus-infected patients were coinfected with bacterial, including Staphylococcus aureus and Streptococcus pneumonia, which resulted in higher morbidity and mortality (9,38,124,125,144). Previous studies using mouse models suggested that influenza A virus infection reduced the number of macrophages and neutrophils in the airway, resulting in the attenuation of innate immune responses against secondary bacterial infection (127). This attenuated innate immune response led to excessive cytokine production caused by secondary infection with S. aureus (75,127). Legionella pneumophila is also a clinically relevant complication of influenza infection (52). In mice infected with influenza A virus, coinfection with L. pneumophila results in increased mortality of mice infected with influenza A virus infection (55). In these mice, coinfection increased the production of proinflammatory cytokines in the lung. However, when TNF-α, MyD88, and TLR2/4 were knocked out, the survival of coinfected mice were not improved, and it is suggested that not inflammation, but a failed tolerance to tissue damage is essential for lethal outcome due to coinfection (55). Indeed, an EGF family member, amphiregulin, which contributes to tissue homeostasis in the lung during influenza A virus infection, decreased lung damage and improved the survival of coinfected mice (55,98). Amphiregulin is produced by innate lymphoid cells (ILCs) and regulatory T cells in the lung (5,98), which are cells known to play a critical role in tissue homeostasis during influenza A virus infection (98). Recent studies reported that EGFR signaling attenuates RIG-I signaling during viral infection (112,160), and amphiregulin is known to promote EGFR signaling (180). Therefore, in addition to tissue repair, amphiregulin might moderately reduce RIG-I signaling to improve survival.
ILCs are classified into three groups, group 1, 2, and 3 (149). Although ILCs contribute to the tissue repair in the lung, their excessive activation is harmful to the host. In addition, ILCs are known to induce type 2 immune responses (170). The group 2 innate lymphoid cells (ILC2s) produce large amounts of IL-5 and IL-13 in response to IL-25 and IL-33 (132,149). IL-5 is a major type 2 cytokine that induces eosinophil production (156), and IL-13 also induces eosinophil recruitment to the lung (121). Excessive expression of IL-33 is a marker of severe and refractory asthma (122). Influenza A virus infection induces the production of IL-33 by alveolar macrophages and NKT cells, which in turn induce IL-5 and IL-13 production by ILC2 (13,40). ILC2-mediated IL-5 production leads to the accumulation of eosinophils in the lung (40).
IL-33 is a ligand of ST2, which induces the production of Th2-associated cytokines (59). Interestingly, it has been reported that cigarette smoke reduces ST2 expression in ILC2, but increases it in macrophages and NK cells, which results in both attenuation of ILC2 function and amplification of type 1 inflammatory responses during viral infection, and as a consequence promotes tissue damage (69). A recent study showed that fine particles induce alveolar macrophage-mediated IL-1α production in the lung, resulting in the promotion of inducible bronchus-associated lymphoid tissue formation (79). Considering that IL-1α is known to induce proliferation of ILC2 and to promote the production of IL-5 and IL-13 (101), it is expected that inhaled particles also affect ILC2-dependent immune responses.
Perspectives
PRRs exert its antiviral effects through production of cytokine and DC-mediated naive T cell priming. A growing body of evidence is accumulating to show that PRRs-mediated innate immune responses are important for controlling viral infection. But, it is well known that host immune responses are sometimes harmful to the host. Allergenic reactions are induced by nonpathogenic molecules, leading to serious diseases, such as anaphylactic shock. Therefore, the immune system is sometimes referred to as a double-edged sword (2,71), and it is applicable to innate immune responses during influenza A virus infection. PRRs are required for controlling influenza A virus replication, but it sometimes causes excessive inflammation. Therefore, loss of PRRs does not lead to the improved survival of mice infected with influenza A virus as described above. In contrast, several recent studies have shown that partial impairment of innate immune responses sometimes successfully improved the survival. However, appropriate control of the innate immune response is still difficult, because environmental factors or secondary infections also affect the innate immune response. In addition, although recent studies revealed the important roles of extracellular vesicles (EVs) for intercellular communications, it is still unclear whether EVs are involved in the innate immune responses during influenza A virus infection.
EVs, including exosomes and microvesicles, are released from multivesicular bodies and from the plasma membrane of host cells, respectively (81). These EVs deliver host and viral proteins as well as nucleic acids from virus-infected cells to DCs and macrophages, thereby regulating innate immune responses (26,73,89,99,102). The role of EVs in the antiviral innate immune response to influenza A virus is elusive, and the effects of EVs on influenza virus pathogenesis remain unclear. Further studies focusing on the role that EVs play in the innate immune response and excessive inflation in the lung are required for a better understanding and for controlling harmful inflammation.
Footnotes
Acknowledgments
This work was supported in part by Grant-in-Aid from the Ministry of Education, Science, and Culture and the Ministry of Health, Labor, and Welfare of Japan, and also supported by Grant-in-Aid from the Japan Agency for Medical Research and Development, PRESTO JST, Mochida Memorial Foundation, Japan Diabetes Foundation, Takeda Science Foundation, the Kao Foundation for Arts and Sciences, and the Japanese Association for Complement Research.
Author Disclosure Statement
No competing financial interests exist.