
Abstract
Notch signaling enhanced the response of interleukin (IL)-22-producing CD4+ T cells that were defined as T helper 22 (Th22) cells, and Notch-aryl hydrocarbon receptor (AhR)-IL-22 axis fine-tuned inflammatory response. Previous studies have demonstrated that both Notch signaling and Th22 cells took part in the pathogenesis of chronic hepatitis C virus (HCV) infection. Thus, in this study, we aimed at examining the regulatory role of Notch signaling in Th22 cells in HCV infection. A total of 59 patients with chronic hepatitis C and 22 normal controls (NCs) were enrolled in this study. The percentage of Th22 cells and mRNA expression of related transcriptional factors and cytokines were analyzed in response to γ-secretase inhibitor. Th22 cell frequency was significantly elevated in chronic hepatitis C in comparison with that in NCs. Inhibition of Notch signaling downregulated HCV-specific Th22 cells and IL-22 production, which was accompanied by the reduction of AhR and modulatory cytokines (IL-6 and tumor necrosis factor-α). Moreover, the suppression of Notch signaling also decreased the IL-22-mediated antimicrobial response in both normal and HCV-infected HepG2 cells/Huh7.5 cells. This process was also accompanied by the depression of signal transducers and activators of transcription 3 signaling. In conclusion, the current results suggested that Notch signaling acted as a critical pathway in determining the response to IL-22 in chronic hepatitis C. Thus, Notch-Th22 axis might be considered a new therapeutic target for HCV-infected patients.
Introduction
C
T helper 22 (Th22) cells represent distinct human CD4+ T cells producing interleukin (IL)-22 and tumor necrosis factor-α (TNF-α), but not expressing IL-17A or interferon-γ (14,15,19,38). IL-22 is the major secreting cytokine by Th22 cells, and it plays dual functions with either pathologic or protective effects in a potential context-dependent manner (3,6,33). There was a robust increase of serum IL-22 in patients with acute hepatitis B, and this elevation has proved to play a proinflammatory role in HBV transgenic mouse models (44). More recent studies also revealed that upregulation of peripheral Th22 and plasma IL-22 reversely correlated with prognosis of HBV-related acute-on-chronic liver failure (29). In contrast, circulating and liver-resident IL-22-producing cells were more hepato-protective and served as reliable predictors for favorable prognosis of drug-induced liver injury (25). In chronic hepatitis C patients, IL-22 was elevated in the serum (9,43) and IL-22-secreting cells were also enriched in the liver (17). The increase of IL-22 triggered hepatic stellate cells, and it regulated liver fibrosis and cirrhosis (36,43) without anti-HCV activity (9).
Notch signaling determined the generation of T and B cell precursors from hematopoietic stem cells (8), and it also biased precursor cells toward a different cell linage in a thymus microenvironment (11). Notch signaling increased IL-22 production in CD4+ T cells by the stimulation of aryl hydrocarbon receptor (AhR), even in the absence of signal transducers and activators of transcription 3 (STAT3). Notch-AhR-IL-22 fine-tuned the immune system and controlled inflammatory responses (1). Furthermore, a recent study demonstrated that Notch signaling regulated IL-22-secreting cells and contributed to liver inflammation in HBV infection (41). Thus, we hypothesized that Notch signaling modulated IL-22-producing CD4+ T cells in HCV infection. To test this possibility, we examined the percentage of Th22 cells and downstream signaling molecules of IL-22 in response to the inhibition of Notch signaling in vitro.
Materials and Methods
Subjects
A total of 59 patients with chronic hepatitis C were enrolled in this study. The diagnosis of chronic hepatitis C was made according to the diagnostic standard of the Chinese National Program for Prevention and Treatment of Viral Hepatitis. All patients were hospitalized or followed up in the Affiliated Hospital to Changchun University of Chinese Medicine from March 2014 to April 2015. No patients were positive for other hepatitis virus or human immunodeficiency virus (HIV) infection, or received antiviral or immunomodulatory therapies before sampling. Patients who were accompanied by liver cirrhosis, liver failure/severe hepatitis, or hepatocellular carcinoma were also excluded. For normal controls (NCs), 22 healthy individuals with matched average age and sex ratio were also enrolled. The characteristics for enrolled subjects are shown in Table 1. The study protocol was approved by the ethics committee of the Affiliated Hospital to Changchun University of Chinese Medicine, and written informed consent was obtained from each subject.
Data are shown as median and range.
ALT, alanine aminotransferase; HCV, hepatitis C virus.
Isolation of peripheral blood mononuclear cells and CD4+ T cells
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque (Sigma, St. Louis, MO) density gradient centrifugation. CD4+ T cells were purified by the human CD4+ T cells isolation kit (Miltenyi Biotech, Bergisch Gladbach, Germany) by following the manufacturer's instructions. The purity of enriched CD4+ T cells was >95% by flow cytometry analysis.
Cell culture
PBMCs or CD4+ T cells were seeded into 24-well plates with the concentration of 1 × 106/mL, and they were cultured in RPMI 1640 that was supplemented with 10% of heat-inactivated fetal bovine serum (FBS) at 37°C under 5% CO2 condition. Cells were stimulated by PMA (final dilution, 50 ng/mL) and ionomycin (final dilution, 1 μg/mL), or HCV Th epitope NS3 1248–1261 (final concentration of 20 μg/mL; sequence: GYKVLVLNPSVAAT) (12,45), in the presence of monensin (final dilution, 10 μg/mL, Sigma). In some experiments, cells were pretreated with γ-secretase inhibitor (GSI) LY-411575 (final concentration 1 μM; Adooq Bioscience, Irvine, CA) for 24 h, and they were washed twice with RPMI1640 for further analysis. HepG2 cells were cultured and grown in Dulbecco's modified Eagle's medium (DMEM) that was supplemented with 10% heat-inactivated FBS, penicillin (100 U/mL), and streptomycin (0.1 mg/mL). The cells were incubated at 37°C under 5% CO2 condition by infection of the serum from HCV RNA positive patients (>107 copies/mL) for 72 h, and HCV RNA levels in HepG2 cells were tested to confirm the infection. The infected HepG2 cells were harvested with GSI stimulation for 24 h, and recombinant IL-22 (final concentration, 1 μg/mL; PeproTech, Rocky Hill, NJ) was also added for another 6 h of incubation.
Generation of HCV viral stocks and infection of Huh7.5 cells
Infectious HCV in cell culture (HCVcc) was generated as previously described (26,40). The pFL-J6/JFH plasmid containing full-length HCV chimeric genome (genotype 2a), and human differentiated hepatocyte-derived cellular carcinoma cell line Huh7.5 cells were kindly provided by Dr. Charles M. Rice (Rockefeller University, New York, NY). HCV RNA was transcribed from pFL-J6/JFH plasmid in vitro by using Ampliscribe T7 High Yield Transcription Kits (Epicentre, Madison, WI) according to the manufacturer's instructions, and it was electroporated into Huh7.5 cells. HCVcc was obtained from cell culture supernatants by precipitation and filtration 8 days post-transfection. The copy number of HCVcc in the cell supernatants was determined by a commercial quantitative reverse-transcription polymerase chain reaction kit (Da'an BioTech, Shenzhen, Guangdong Province, China). Huh7.5 cells were seeded into a 24-well plate, and 1 × 107 copies of HCVcc were added and incubated for 5 h. Infected cells were washed with DMEM for five times, and they were supplemented with fresh DMEM that was supplemented with 10% heat-inactivated FBS, penicillin (100 U/mL), and streptomycin (0.1 mg/mL). Infected Huh7.5 cells were cultured for 48 h, and HCV RNA levels in Huh7.5 cells were tested to confirm the infection. The infected Huh7.5 cells were then stimulated with GSI for 24 h, and recombinant IL-22 (final concentration, 1 μg/mL; PeproTech, Rocky Hill, NJ) was also added for another 6 h of incubation.
Flow cytometry
Cells were transferred to 5 mL FACS tubes, and anti-CD3-APC (BD Bioscience, San Jose, CA) and anti-CD4-PE (BD Bioscience) were added for a 30 min incubation in the dark at 4°C. Cells were fixed by adding 100 μL of Fixation & Permeabilization Medium A (Caltag Laboratories, Invitrogen, CA) and incubated in the dark at room temperature for 15 min after surface staining. Cells were then resuspended in 100 μL of Fixation & Permeabilization Medium B (Caltag Laboratories) containing anti-IL-17A FITC (BD Bioscience) and anti-IL-22 PerCP (BD Bioscience) for a 20 min incubation. Isotype control antibodies were used to separate positive and negative cells in the APC, PE, FITC, and PerCP fluorescence channels. Samples were analyzed by using a four-color FACS Calibur analyzer (BD Biosciences Immunocytometry Systems, San Jose, CA). Acquisitions were performed with CellQuest Pro software (BD Biosciences Immunocytometry Systems), and analyses were performed with FlowJo version 8.7.2 for Windows (Tree Star, Inc., Ashland, OR).
Real-time reverse polymerase chain reaction
Total RNA was isolated from cultured cells by using RNeasy mini kit (Qiagen, Hilden, Germany) by following the manufacturer's instructions. cDNA was synthesized with random hexamers by using PrimeScript RT Master Mix (TaKaRa, Dalian, China). Real-time PCR was performed by using SYBR Premix Ex Taq (TaKaRa). The relative gene expression was quantified by the ΔΔCT method by using 7500 System Sequence Detection software (Applied Biosystems, Foster, CA). The sequences of the primers were cited from previous studies (7,30,41) and are shown in Table 2.
Enzyme-linked immunosorbent assay
The levels of IL-17A and IL-22 were measured by commercial enzyme-linked immunosorbent assay (ELISA) kits (eBioscience, San Deigo, CA) according to the manufacturer's instructions.
Western blot
HepG2 cells were lysed on ice for 15 min in lysis buffer, and the supernatants were collected by centrifugation at 12,000 g for 10 min at 4°C. The total protein was loaded on SDS-PAGE by using Mini-protean 3 electrophoresis cell systems (Bio-Rad, Hercules, CA), and it was electroblotted onto a PVDF membrane. The membrane was soaked in block solution (PBS containing 10% nonfat milk and 0.05% Tween 20), and then it was incubated overnight in the presence of rabbit anti-phosphorylated STAT3 (phospho Y705) antibody or rabbit anti-STAT3 antibody (1: 1,000 dilution; Abcam, Cambridge, MA). After incubation, the membrane was washed three times, and horseradish peroxidase-conjugated polyclonal goat anti-rabbit antibody (1:2,000 dilution; Abcam) was added for an additional 2 h of incubation. Antibody-antigen complexes were visualized by enhanced chemiluminescence (Western Blotting Luminol Reagent, Santa Cruz, CA).
Statistical analysis
All data were analyzed by using SPSS version 19.0 for windows software (Chicago, IL). Student t-test was used for the comparison between two groups. SNK-q test was used for the comparison among groups. Paired t-test was used for the comparison between before and post-treatment. p-Values of less than 0.05 were considered to indicate significant differences.
Results
The percentage of circulating Th22 and Th17 cells was elevated in patients with chronic hepatitis C
We examined peripheral blood from volunteers to determine the percentage of IL-22-producting CD4+ T (Th22) cells and IL-17-producting CD4+ T (Th17) cells in the total CD4+ T cell population. The typical PBMC samples from patients with both chronic hepatitis C and NC analyzed by flow cytometry are shown in Figure 1A. On the basis of intracellular cytokine staining analyses, little IL-22 or IL-17 production can be found in CD4+ T cells without stimulation. After challenge with PMA and ionomycin for 12 h, the percentages of Th22 cells (phenotype: CD3+CD4+IL-22+IL-17−) and Th17 cells (phenotype: CD3+CD4+IL-22−IL-17+) from hepatitis C patients were significantly higher than those from NCs (Th22: 1.31% ± 0.44% vs. 0.50% ± 0.13%, Student t-test, p = 0.013, Fig. 1B; Th17: 2.16% ± 0.28% vs. 1.55% ± 0.08%, Student t-test, p = 0.0059, Fig. 1C). Moreover, IL-22 and IL-17 expression in the supernatants from cultured PBMCs with PMA and ionomycin stimulation was also measured. Both IL-22 and IL-17 concentrations in cultured PBMCs from HCV-infected patients were remarkably higher than those from NCs (IL-22: 2011 ± 253.8 pg/mL vs. 667.1 ± 187.9 pg/mL, Student t-test, p = 0.0001, Fig. 1D; IL-17: 1437 ± 345.5 pg/mL vs. 361.1 ± 153.9 pg/mL, Student t-test, p = 0.0013, Fig. 1E). Furthermore, the viral-specific Th22 and Th17 cells were also induced by HCV NS3 Th epitope peptide stimulation in HCV-infected patients. As shown in Figure 1F and G, little IL-22 or IL-17 production was found in NCs with NS3 peptide stimulation. HCV-specific Th22 and Th17 cells could be detected from chronic hepatitis C patients. However, there were no remarkable differences in viral-specific Th22 cells (1.44% ± 0.40% vs. 1.34% ± 0.53%, Student t-test, p = 0.821, Fig. 1F) and viral-specific Th17 cells (2.15% ± 0.23% vs. 2.17% ± 0.43%, Student t-test, p = 0.959, Fig. 1G) between HCV genotype 1b- and HCV genotype 2a-infected patients. Unfortunately, we did not find significant correlations between Th22/Th17 cells and virological/biochemical index (HCV RNA/alanine aminotransferase [ALT]) in patients with chronic hepatitis C (p > 0.05, data not shown).

The percentage of Th22/Th17 cells and IL-22/IL-17 production were increased in HCV-infected patients.
Notch1 and Notch2 expression was also elevated in patients with chronic hepatitis C
mRNA expression of Notch1 and Notch2 was measured by real-time reverse polymerase chain reaction (RT-PCR) in PBMCs and CD4+ T cells. Both Notch1 and Notch2 revealed approximately a three-fold induction in PBMCs isolated from chronic hepatitis C in comparison with those in NCs (Student t-test, p = 0.032 and p = 0.0016, respectively, Fig. 2A, B). Furthermore, more than 10-fold elevations of Notch1 and Notch2 were found in CD4+ T cells that were isolated from chronic hepatitis C in comparison with those in NCs (Student t-test, p < 0.0001, Fig. 2C, D).
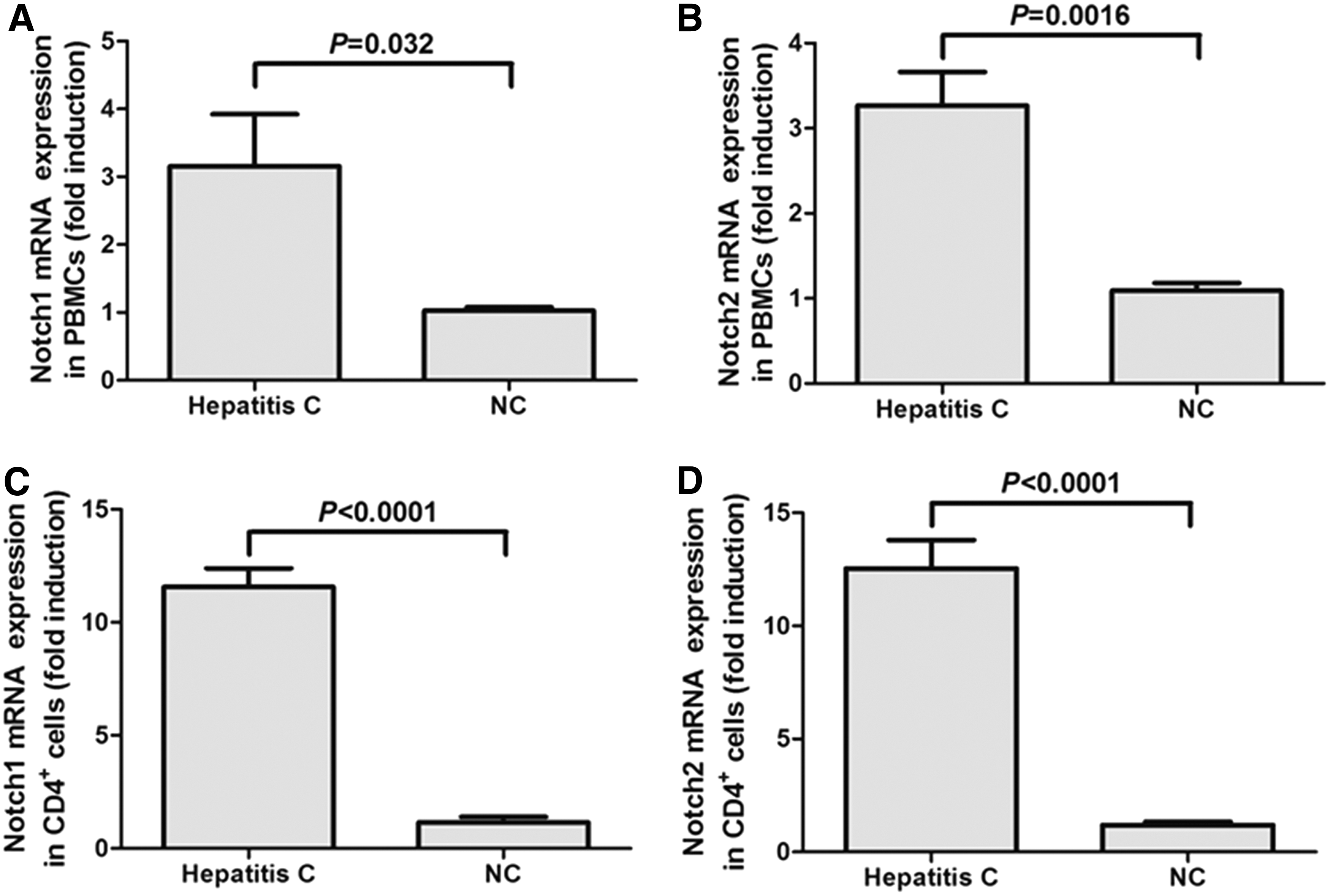
Notch1 and Notch2 mRNA expression was elevated in both PBMCs and CD4+ T cells from chronic hepatitis C patients. Notch1 mRNA in PBMCs
Inhibition of Notch signaling reduced frequency of HCV-specific Th22 cells
The purified CD4+ T cells from 23 patients of chronic hepatitis C were cultured with GSI for 24 h. Cells were washed twice with RPMI 1640, and they were then stimulated by HCV NS3 Th epitope peptide with monensin for 12 h, or without monensin for 48 h. Cells with monensin treatment were harvested for flow cytometry analysis. Cells without monensin treatment were harvested for RT-PCR assay, whereas the supernatants were also maintained for cytokine assay by ELISA. Notch signaling inhibition was confirmed by downregulation of Hes1 and Hes5 mRNA expression in response to GSI stimulation (Fig. 3A, B). Inhibition of Notch signaling by GSI notably reduced the percentage of viral-specific Th22 cells within CD4+ T cells (1.72% ± 0.32% vs. 1.01% ± 0.12%, paired t-test, p = 0.036, Fig. 3C). There was also a consistent trend of downregulation in HCV-specific Th17 cells in response to GSI stimulation. However, this difference failed to achieve statistical significance (2.64% ± 0.72% vs. 2.07% ± 0.39%, paired t-test, p = 0.228, Fig. 3D). The secreting cytokines by Th22 and Th17 cells in the supernatants were also measured by ELISA. The concentrations of IL-22 and IL-17 revealed similar trends, with the changes of Th22 and Th17 cells in response to Notch signaling inhibition. GSI stimulation induced the reduction of both viral-specific secreting cytokines. However, only the downregulation of IL-22 demonstrated a statistical difference (718.2 ± 271.9 pg/mL vs. 189.5 ± 69.70 pg/mL, Paired t-test, p = 0.0074, Fig. 3E), whereas there were no remarkable changes of IL-17 levels with the suppression of Notch signaling (340.4 ± 122.9 pg/mL vs. 222.8 ± 104.2 pg/mL, Paired t-test, p = 0.089, Fig. 3F).
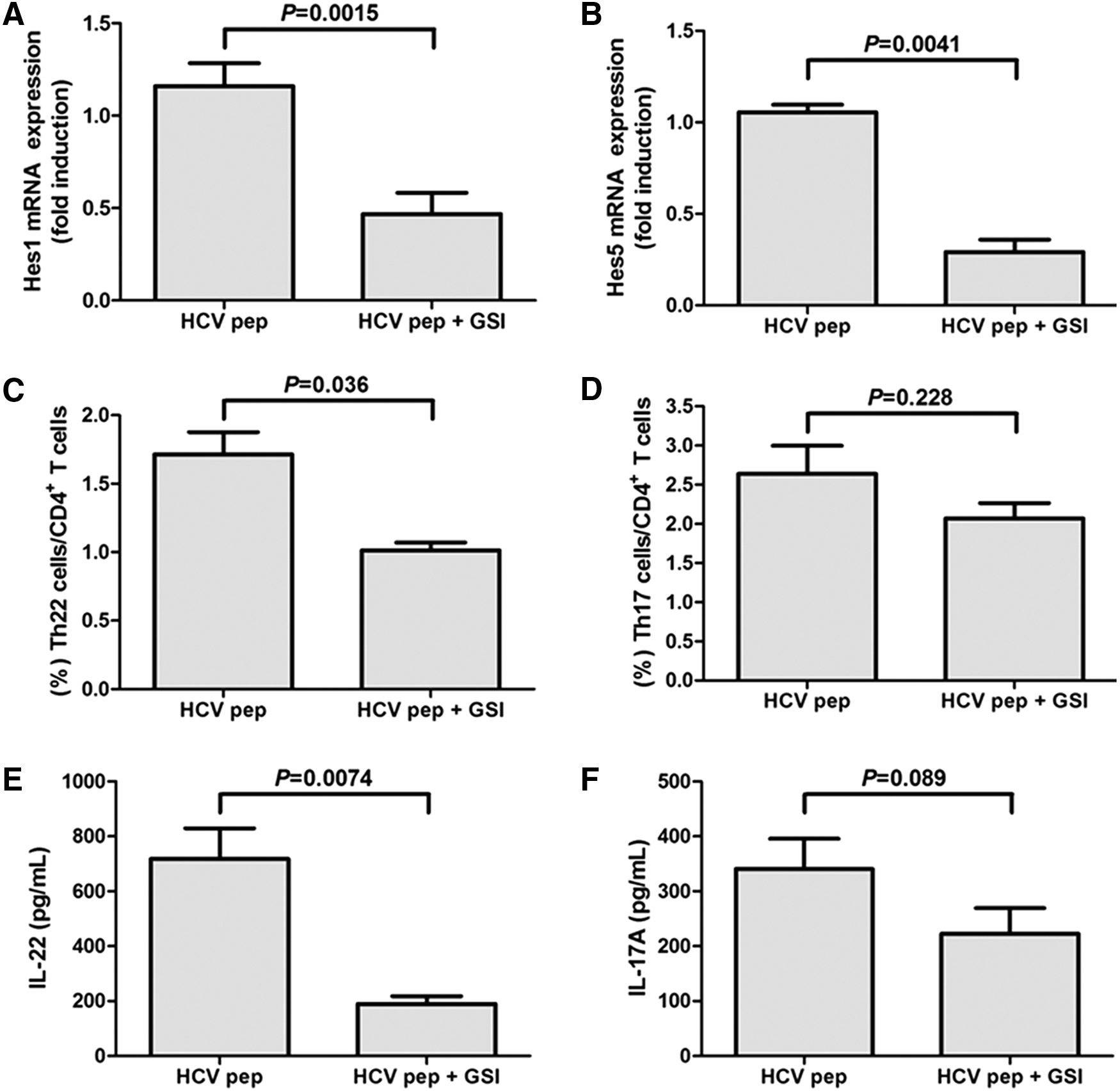
Inhibition of Notch signaling reduced Th22 cell frequency and IL-22 production in purified CD4+ T cells from chronic hepatitis C. Isolated CD4+ T cells from chronic hepatitis C were stimulated by HCV NS3 Th peptide, with or without γ-secretase inhibitor (GSI) LY-411575. The downregulations of Hes1
Inhibition of Notch signaling decreased AhR mRNA expression in HCV-infected patients
The mRNA expression of transcriptional factors that were associated with Th22 and Th17 cells was measured by RT-PCR. Th22-related transcriptional factor AhR was elevated in response to HCV peptide stimulation (SNK-q test, p = 0.0002, Fig. 4A). GSI significantly decreased HCV-induced AhR levels only slightly higher than mock stimulation (SNK-q test, p = 0.0006, Fig. 4A). However, viral peptide-induced Th17-related transcriptional factor retinoid-related orphan receptor γt (RORγt) was not remarkably reduced in GSI-treated CD4+ T cells in comparison with untreated cells (SNK-q test, p = 0.614, Fig. 4B). Furthermore, the related cytokine mRNA levels of Th22 and Th17 cells were also investigated. Both IL-6 and TNF-α mRNA level was increased with HCV peptide stimulation (SNK-q test, p = 0.0018, Fig. 4C; p = 0.0014, Fig. 4D), but it decreased in the presence of Notch inhibitor (SNK-q test, p = 0.0039, Fig. 4C; p = 0.032, Fig. 4D). mRNA level corresponding to IL-23 did not change notably between GSI-treated and -untreated CD4+ T cells (Fig. 4E).
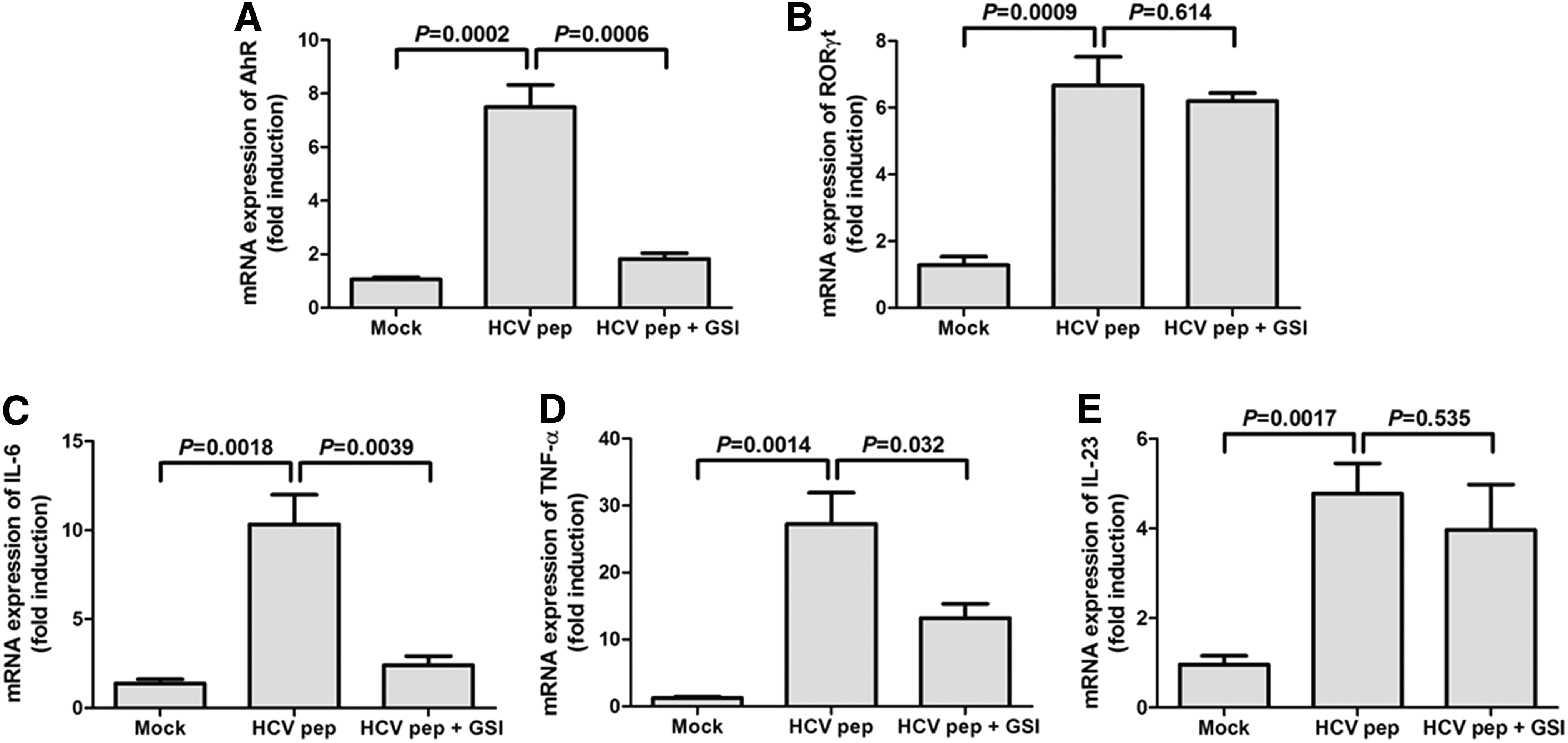
The mRNA expression of transcriptional factors and related cytokines was analyzed by RT-PCR. Isolated CD4+ T cells from chronic hepatitis C were stimulated by HCV NS3 Th peptide, with or without GSI LY-411575. The mRNA expression of aryl hydrocarbon receptor (AhR)
IL-22-mediated induction of REG family genes could be suppressed by inhibition of Notch signaling in HCV-infected HepG2 cells/Huh7.5 cells
A previous study demonstrated that IL-22-STAT3 signaling increased the induction of genes encoding antimicrobial peptides, including REG1A, REG3A, and REG3G. Inhibition of Notch signaling reduced the induction of those genes, leading to a poor response to IL-22 in intestinal epithelial cells (30). To confirm the role of Notch-IL-22 axis in HCV infection, both normal and HCV-infected HepG2 cells were treated with GSI for 24 h, and then stimulated with IL-22 for the next 6 h. HCV infection seemed to induce phosphorylated STAT3, because no p-STAT3 could be found in normal HepG2 cells but a low level of p-STAT3 could be tested in HCV-infected HepG2 cells (Fig. 5A). GSI decreased the IL-22-induced phosphorylation of STAT3 in both normal and HCV-infected HepG2 cells; however, the total STAT3 did not change significantly in response to GSI (Fig. 5A). Moreover, treatment with GSI also significantly downregulated the IL-22-mediated induction of REG1A (SNK-q test, p = 0.026, Fig. 5B), REG3A (SNK-q test, p = 0.0058, Fig. 5C), and REG3G (SNK-q test, p = 0.01, Fig. 5D) in HCV-infected HepG2 cells. The changes of REG1A, REG3A, and REG3G revealed similar trends in normal HepG2 cells with IL-22 and GSI stimulation (Fig. 5B–D).

IL-22-mediated induction of REG family genes suppressed by the inhibition of Notch signaling in both normal and HCV-infected HepG2 cells. Normal and HCV-infected HepG2 cells were harvested with GSI LY-411575 stimulation for 24 h, and recombinant IL-22 was also added for another 6 h of incubation.
To further confirm the role of Notch-IL-22-induced antimicrobial peptide expression in vitro, we used the HCVcc-infected Huh7.5 cells to again perform the experiments mentioned earlier. As shown in Figure 6, GSI stimulation also remarkably reduced the IL-22-induced expression of REG1A (SNK-q test, p = 0.021, Fig. 6A), REG3A (SNK-q test, p = 0.032, Fig. 6B), and REG3G (SNK-q test, p = 0.0022, Fig. 6C) in HCVcc-infected Huh7.5 cells. The changes of REG1A, REG3A, and REG3G revealed similar trends in normal Huh7.5 cells with IL-22 and GSI stimulation; however, IL-22-mediated RGE3G did not reduce significantly in response to GSI treatments in normal Huh7.5 cells.
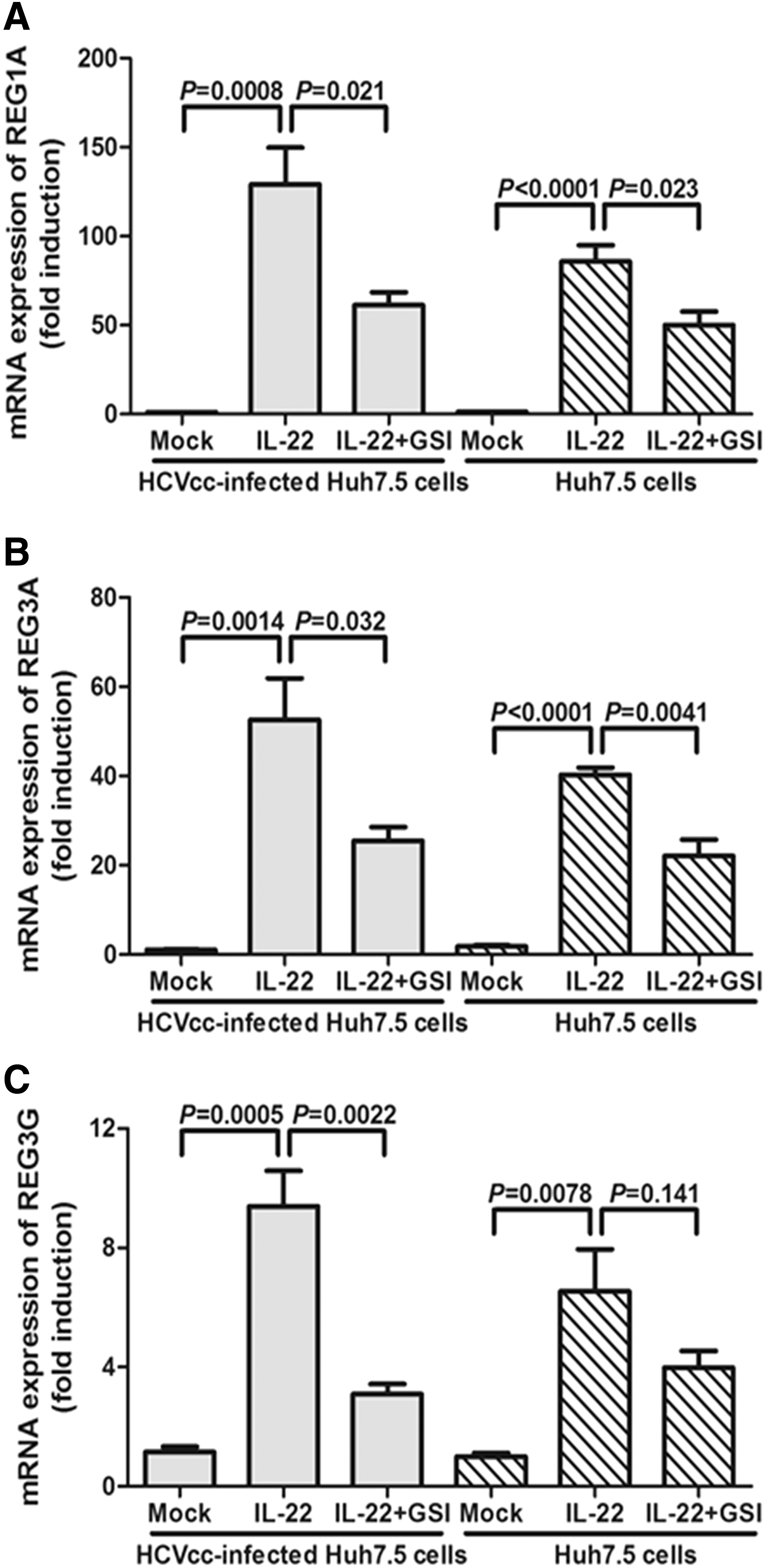
IL-22-mediated induction of REG family genes suppressed by the inhibition of Notch signaling in HCVcc-infected Huh7.5 cells. Normal and HCVcc-infected Huh7.5 cells were harvested with GSI LY-411575 stimulation for 24 h, and recombinant IL-22 was also added for another 6 h of incubation. GSI significantly downregulated the IL-22-mediated induction of REG1A
Discussion
In the present study, we demonstrated the elevation of both Th17 and Th22 cells in chronic hepatitis C in comparison with those in healthy individuals. Inhibition of Notch signaling downregulated HCV-specific Th22 cells and IL-22 production, without affecting Th17 cells and IL-17A secretion. This process was accompanied by the reduction of transcriptional factor AhR and modulatory cytokines IL-6 and TNF-α, but not RORγt or IL-23. Furthermore, the suppression of Notch signaling also decreased the IL-22-mediated antimicrobial response partly by depressing STAT3 signaling. Our current findings provided the role of Notch-Th22 axis in the regulation of chronic HCV infection.
Th22 cells have been revealed to play vital roles in the pathogenesis of both acute and chronic viral infections. Th22 cells, as well as associated cytokines and transcriptional factors, were significantly increased in patients with enterovirus 71 virus-infected hand, foot, and mouse disease, particularly in severe patients (7). In contrast, HIV infection induced the downregulation of circulating and mucosal Th22 cells and IL-22 production, whereas antiretroviral therapy restored Th22 activity in gut mucosa integrity (16,22,31). Furthermore, the increased frequency of Th22 cells played an important role in the pathogenesis of coxsackie virus B3-induced mouse acute or chronic viral myocarditis, and IL-22 might be a myocardium-protective cytokine by the inhibition of myocardial fibrosis (20,24). Consistent with the previous findings (23), the present results indicated an increase in both viral-specific and -nonspecific Th22 cells, but not Th17 cells, in chronic HCV infection. However, this elevation did not closely correlate with either HCV RNA or ALT level. It is well accepted that IL-22 has minimal function in the suppression of viral replication (32,44). Due to the dual nature of Th22 and secreting IL-22 in various diseases, which possibly aggravate or attenuate disease severity, the potential role of Th22 cells and related cytokines in patients with chronic hepatitis C is not yet known.
The regulation of Th22 cells in the progression of diseases was not well understood. It has been widely known that IL-6 and TNF-α promoted the Th22 phenotype with the assistance of plasmacytoid dendritic cells (15,27,37). A recent study indicated that runt-related transcription factor 3 (RUNX3) is involved in the differentiation of Th17 and Th22 cells in psoriasis (18). Importantly, Notch signaling enhanced IL-22-secreting CD4+ T cells, which are partly dependent on AhR stimulation in the ConA-mediated acute hepatitis mouse model (1). Moreover, Notch signaling also increased the function of NKp46+ innate lymphoid cells 22 (ILC22), which were also one of the major sources of IL-22, in the HBV-infected mouse model (41). However, the inhibition of Notch signaling did not affect CD4+RORγt+ cells (mainly Th17 cells) in HBV infection (41). This was consistent with the current findings in HCV infection. However, a more recent study by Qin et al. on the role of Notch signaling in balancing regulatory T cells and Th17 cells showed that the inhibition of Notch signaling reduced Th17 cells (35). We found that Notch signaling did not influence Th17 cell responses. It was probably because Qin et al. tested nonspecific Th17 cells but we measured HCV-specific Th17 cells. Moreover, Notch signaling regulated viral-specific Th22 cells as well as the expression of secreting cytokine IL-22 and transcriptional factor AhR, indicating that Notch-AhR-IL-22 axis contributed to chronic HCV infection. Furthermore, the suppression of Notch signaling also reduced HCV peptide-induced IL-6 and TNF-α expression. Th22 cells were known to produce TNF-α (15,19), and, in turn, TNF-α promoted Th22 proliferation and differentiation (27). Thus, Notch signaling might increase this positive feedback function of TNF-α, and subsequently, it regulated viral-specific Th22 cells in patients with chronic hepatitis C.
Signaling through IL-22-IL-22 receptor (IL-22R) always resulted in the phosphorylation of STAT3, leading to a variety of proliferative, anti-apoptotic, and anti-microbial pathways (13). Since IL-22R was restrictedly expressed in the tissue but not immune cells, we used the HCV-infected hepatocellular carcinoma cell line HepG2 and HCVcc-infected Huh7.5 cells for further analysis. Activation by IL-22 induced high-level expression of p-STAT3, whereas inhibition of Notch signaling reduced the phosphorylation of STAT3. Subsequently, IL-22 also enhanced the expression of antimicrobial genes, especially REG family genes. Moreover, IL-22-mediated induction of REG1A, REG3A, and REG3G was also regulated by Notch signaling. This was consistent with the previous study in human intestinal epithelial cells (30). REG1A and REG3A could be induced in HBV- or HCV-infected no-tumor hepatitis/cirrhotic tissues (39). Strong upregulation of REG3G was also found after influenza virus infection; however, REG3G gene had no strong effect on the susceptibility of the host to infections with influenza virus in the mouse model (42). REG family genes were regulated by STAT3 and highly elevated after Methicillin-resistant Staphylococcus aureus infection in lung epithelium (4) and Listeria monocytogenes infection in the intestinal tract (2). Thus, IL-22-target REG family genes encoding antimicrobial peptides effectively contributed to bacterial and parasitic infection, but they have minimal function in controlling viral infection. This might partly explain the reason that IL-22 has little effect in suppressing viral replication.
Conclusion
In summary, our results showed that the Notch signaling regulated the HCV-induced elevations of Th22 cell frequency, associated cytokines (IL-22, TNF-α, and IL-6), and transcriptional factor AhR expression. The current findings suggested that Notch signaling acted as a critical pathway in determining the response to IL-22 in chronic hepatitis C. Therefore, Notch-Th22 axis might be considered a new therapeutic target for HCV-infected patients.
Footnotes
Acknowledgments
The authors thank Dr. Xin Wei (Tangdu Hospital) for performing the HCVcc experiments, and Dr. Charles M. Rice (Rockefeller University) for providing Huh7.5 cells and the pFL-J6/JFH plasmid.
Author Disclosure Statement
No competing financial interests exist.