
Abstract
The lung is an important line of defense that is exposed to respiratory infectious pathogens, including viruses. Lung epithelial cells and/or alveolar macrophages are initially targeted by respiratory viruses. Once respiratory viruses invade the cells of the lung, innate immunity is activated to inhibit viral replication. Innate immune signaling also activates virus-specific adaptive immune responses. The helper T cells play pivotal roles in the humoral and cellular adaptive immune responses. Helper T cells are categorized into several distinct subsets (e.g., TH1, TH2, TFH, TH17, and Treg), differentiated by their corresponding signature cytokine production profiles. Helper T cells migrate into the airways and the lung after respiratory virus infections. The behavior of the helper T cells differs with each respiratory virus—in some cases, the response is beneficial; in other cases, it is harmful. Here, the general mechanisms underlying helper T cell responses to viral infections are summarized, and functions and reactions of the helper T cells against some respiratory viral infections are discussed. In influenza virus infections, TH1 cells, which regulate the cytotoxic T lymphocytes and IgG2 responses, are efficiently activated. TFH cells required for highly specific and memory humoral responses are also activated on influenza infections. In infections with respiratory syncytial virus and rhinovirus, TH2 cells develop in the lung and contribute to pathogenesis. In many cases, Treg cells inhibit excessive virus-specific T cell responses that can contribute to viral pathogenicity.
Introduction
R
Respiratory viruses invade the body through the nasal cavity and throat, and rapidly replicate initially in the local epithelium. Then, viruses spread into airways and lungs. The majority of respiratory viruses are lytic viruses, which kill the infected host cells on viral release therefrom. Lungs have specialized epithelial cells and sophisticated vascular networks to achieve efficient gas exchange. Cell death due to viral replication can compromise the respiratory function and cause pulmonary edema, and sometimes severe pneumonia. In most severe cases, the effects of viral replication are not limited to the respiratory tract, and also become systemic. Abnormal systemic elevation of cytokines and chemokines, called a cytokine storm and caused by the breakdown of pulmonary epithelium and vascular endothelium, causes dysfunction of other organs (e.g., liver and brain) (123).
One to three days after a viral infection of the upper respiratory tract, dendritic cells (DCs) capture the viral antigens and migrate into secondary lymphoid organs. Naive CD4+ T cells in the T cell zone of the secondary lymphoid organs are involved in antigen presentation by DCs and develop into each of the helper T cell subsets within 3 to 6 days. Subsets of the helper T cells are defined by the type of cytokines they produce (Fig. 1). The major helper T cell subsets are TH1, TH2, TFH, TH17, and Treg, which develop depending on the environment and the magnitude of T cell receptor (TCR) signaling (139). Among these subsets, TH1 cells become the most abundant in the respiratory tract after influenza virus infection. Viral compartments and tissue damage caused by viral replication skew the environment toward the development of TH1 cells. TH1 cells are mainly thought to contribute to CTL responses; however, we recently showed that TH1 cells play a significant role in the protective humoral responses in the context of vaccination against influenza virus (86).
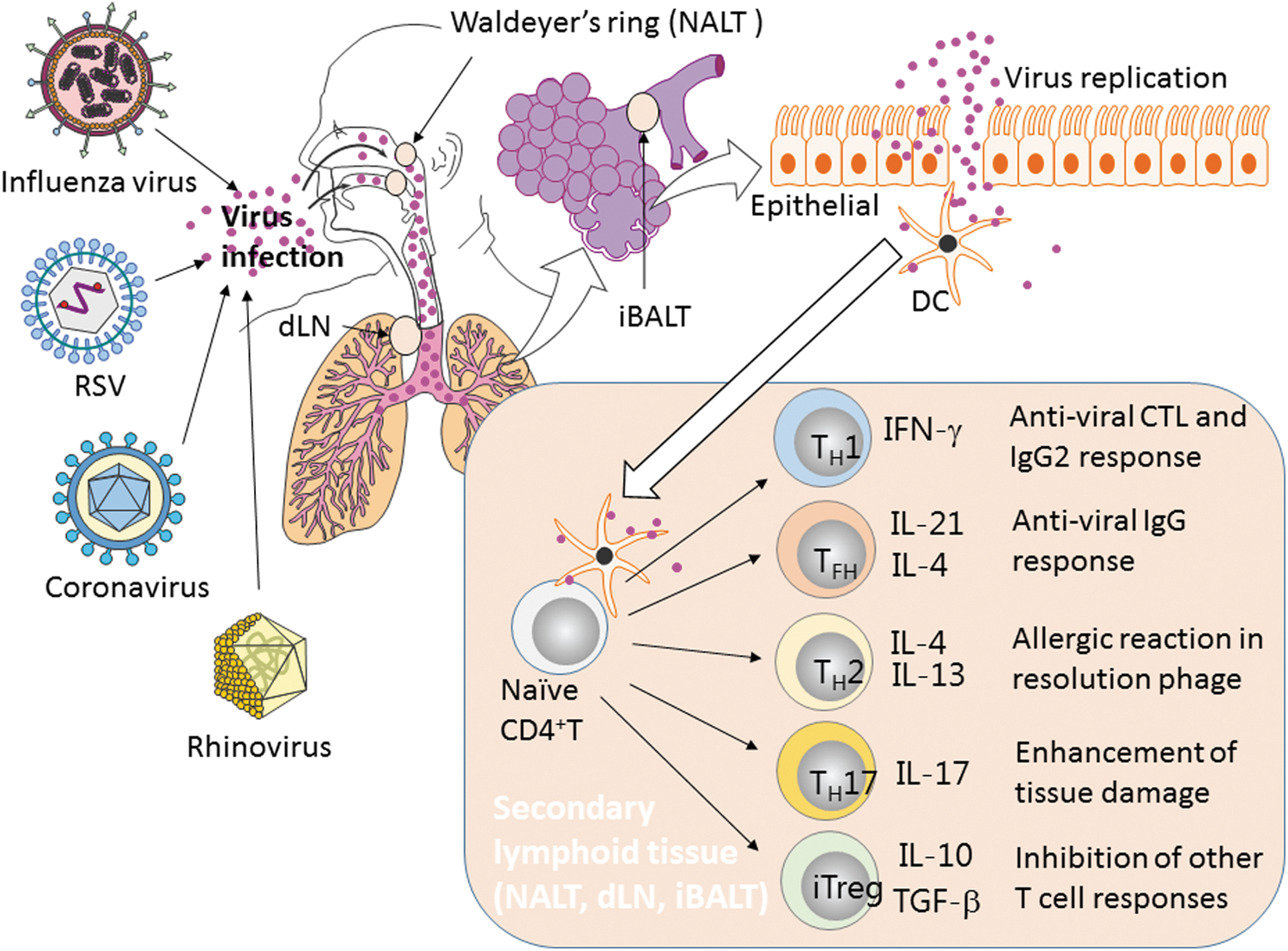
Helper T cell induction and functions to respiratory viruses in the lung. After respiratory viral infection of the upper respiratory tract, DCs capture the viral antigens and migrate into secondary lymphoid organs. Naive CD4+ T cells in the T cell zone of the secondary lymphoid organs are activated by DCs and differentiate into each of the helper T cell subsets. Each subset of the helper T cells exhibits a function through the cytokine production. CTL, cytotoxic T lymphocytes; DCs, dendritic cells; dLN, draining lymph nodes; iBALT, induced bronchus-associated lymphoid tissue; IFN, interferon; IL, interleukin; NALT, nasal-associated lymphoid tissue; RSV, respiratory syncytial virus; TGF-β, transforming growth factor-beta.
Another major cell subset involved in the viral immune responses is TFH, specialized helper T cells involved in B cell activation and antigen production (28). The germinal center (GC) is an important structure for selection and clonal expansion of antigen-specific B cells. TFH cells stay in the GC and select B cells, which have high affinity for antigens, by interacting with cognate B cells. The GC is formed within the B cell follicle in secondary lymphoid tissues, which consist of the T cell zones and B cell follicles. Nasal-associated lymphoid tissue (NALT), bronchus-associated lymphoid tissue (BALT), and mediastinal lymph nodes (lung-draining lymph nodes) are secondary lymphoid tissues of the respiratory tract (37,47,56). NALT is located behind the nasal cavity in rodents. Waldeyer's tonsillar ring has been speculated to function as NALT in humans (53). BALT exists in the airways of guinea pigs and rabbits. Induced BALT (iBALT), resembling the structure of BALT, is formed near the blood vessels in the lung after respiratory viral infections in humans and mice (88,103). These lymphoid tissues can be a major site for helper T cell development and activation.
Influenza virus is the most well-studied virus in regard to the helper T cell responses in the respiratory tract. Development of all of the major subsets of helper T cells has been reported during influenza infection, although their functions and physiological relevance are controversial. The second most studied respiratory infectious virus is respiratory syncytial virus (RSV), which infects mostly children before the age of two and causes severe lung disorder in some cases (133). Although both influenza virus and RSV infect the lung epithelium, helper T cells react significantly differently in response to different infections. In general, RSV has a greater ability to control the helper T cell functions than influenza virus can. In this review, first, the general concept of reactions of the helper T cells during respiratory viral infections is discussed. Later, unique responses of the helper T cells against some respiratory viruses are detailed. A deeper understanding of the helper T cell responses to infectious respiratory viruses is beneficial for understanding pathogenesis and developing novel vaccine strategies.
Bridging Innate and Acquired Immune Responses Against Respiratory Viral Infections
Lungs are responsible for gas exchange and are exposed to the external environment. Alveolar cells consist of the lung epithelium and alveolar macrophages, which are targets of respiratory viruses. In the primary infection by a respiratory virus, innate immune responses play a major role in inhibiting viral replication. In cases of influenza virus and RSV infections, virus entry is detected by viral-RNA-sensing systems, including RIG-I and Mx (43,55,101). Interferon (IFN) signaling, which is the most potent system to tackle viral infections, is activated by viral-RNA-sensing systems. IFN families are divided into three types—type I IFN family consists of IFN-α and IFN-β, type II IFN family is only IFN-γ, and type III IFN family consists of IFN-λ (108). Type I IFN and type III IFN families have redundant effects in preventing influenza virus replication (27). Type III IFN family also inhibits replication of RSV and corona virus in respiratory epithelial cells (61,87,94). IFN-γ plays significant roles in natural killer cell function and development of virus-specific acquired immunity. Viral-RNA-sensing systems also induce release of various chemokines that attract inflammatory macrophages and neutrophils into lungs' parenchyma. Macrophages and neutrophils clear dead cells and viruses from the lungs (50). During a secondary infection with the same respiratory virus, acquired immunity responds extremely powerfully to prevent the virus spread.
DCs bridge innate and acquired immunity together. Lung residential DCs are activated by innate immunity signaling by thymic stromal lymphopoietin (TSLP) and IL-1 family of cytokines, including IL-18 and IL-33 (58,74,129). Recent studies have indicated that innate lymphoid cells (ILC2) activated by IL-33 and/or TSLP have a role in DC activation (46). Activated DCs engulf the viral antigen and migrate to lung-draining lymph nodes (dLN). Two major DC subsets, CD11b+ and CD11b− populations, exist in the uninfected lung, and the monocyte-derived DC subset develops in the inflamed lung during virus infection (65). Each DC subset is distinguished from the others by cytokine production and major histocompatibility complex (MHC)-II expression, which determine the subset of effector CD4+ T cells (31,90). DCs can be targeted by influenza virus and RSV (48,113). In the case of RSV, infection of DCs may contribute to suppression of helper T cell activation (30).
In general, the detection of respiratory viral infections by virus-sensing systems induces release of chemokines to promote migration of naive T cells, activates IFN signaling, and causes DCs to convert to effector T cells.
Helper T Cell Functions During Antiviral Humoral Responses in the Respiratory Tract
IgG is the major class of virus-specific antibodies produced during systemic humoral responses. On the contrary, IgA antibodies exert significant roles in mucosal humoral responses in the airways (100). The mechanisms underlying systemic virus-specific IgG induction are relatively clearer than those underlying mucosal IgA responses. Affinity maturation of antibodies is achieved by somatic hypermutations in the immunoglobulin genes during selection and clonal expansion of B cells in the GCs, sites of interaction with cognate T cells. TFH cells are specialized helper T cells that support B cell selection in the GC (28). High IL-6 and IL-21 levels and low IL-2 levels augment differentiation of the naive CD4+ T cells into TFH cells (33,92,93,119,126). Mice deficient in T cell-specific Bcl6, a master regulator of TFH cells, do not have TFH cells; this results in the absence of GC in these mice. Bcl6 is a transcriptional repressor that represses development of the TH1 and TH17 cells to skew T cell differentiation toward TFH cells (137). CXCR5, a CXCL13 receptor, is a cellular TFH marker required to retain TFH cells within the B cell follicles (52,97). Signature cytokines produced by TFH cells are IL-21 and IL-4, which cause B cells to switch class in the GC (106). GC and TFH cells develop in the iBALT and dLN that are observed after influenza virus infection (88).
Influenza infection efficiently induces development of the TFH cells (3,11), which interact with cognate B cells in GCs. Mice have four IgG subclasses: IgG1, IgG2b, IgG2a, or IgG2c, depending on the strain, and IgG3. During cognate T–B cell interactions, certain cytokines cause switching of the IgM to IgG subclass. Cytokines support B cell differentiation and proliferation in a manner intrinsic to B cells (140). IL-4 and IL-21 produced by TFH cells induce class switching to IgG1. On the contrary, IFN-γ produced by TFH and TH1 cells induces class switching to IgG2 (114), which is a major subclass of virus-specific IgGs in the serum of immunized mice. High-affinity IgG2 produced by B cells selected in the GC requires the support of TFH cells. However, mice lacking the GC–TFH program retain the virus-specific IgG2 subclass, protecting them from lethal viral challenges, indicating that low-affinity IgG2 induced by TH1 cells is responsible for antiviral IgG responses, and suggesting that TH1 cells also play significant roles during protective humoral responses (86).
Mechanisms underlying induction of antiviral IgA responses in the respiratory tract are still unknown. Although IgA is an abundant Ig class in the serum, respiratory virus-specific IgA is minimal or undetectable in the serum (11). This clearly indicates that systemic and local IgA responses are distinctly regulated. In the case of antibacterial IgA induction in the gut, IgA class switching is regulated by various environmental factors, including T cell-dependent and T cell-independent mechanisms. T cell-dependent IgA induction mainly relies on the GC–TFH cells in the Peyer's patch (100). In the respiratory tract, the GC–TFH dependency for IgA responses has been unknown. Low frequency of TFH cells in NALT and the lung is also enigmatic (11). NALT, iBALT, and dLN are candidate sites for generation of IgA-producing B cells. Lymphotoxin-alpha-deficient mice lack secondary lymphoid tissues (only NALT is retained but its structure is disturbed) (37), but have normal IgA in the respiratory tract (79). Thus, although IgA-producing plasma cells are observed in the lymphoid tissues, the origin and mechanism of IgA production in the airway and lung require further investigation.
TH2 cells have a significant role in the production of IgE and protection against helminth parasites (41,111). TH2 cells produce IL-4, IL-5, and IL-13 as signature cytokines that enhance IgE production and migration of eosinophils, basophils, and additional TH2 cells (134). TH2 cells are tightly associated with asthma during the resolution phase of influenza infection (105).
iBALT has typical B cell and T cell zones that are rich in CCL21 and CXCL13, CCR7 and CXCR5 ligands, respectively (88). CXCR5 is a major signature molecule for TFH cells; however, lack of CXCL13 does not affect iBALT structure (104). TNF-α and IL-17 mediate the development of iBALT (103). IL-6 is also required for TFH differentiation (28,92). These cytokines are abundant in the lung after influenza infection. Efficient secondary IgG responses by memory B cells and long-lived plasma cells depend on the iBALT-associated GC (99). Production of antiviral IgG with cross-reactivity also requires pulmonary GCs (1).
Both systemic IgG and local IgA humoral antiviral responses are regulated by the helper T cells that are induced by respiratory viral infections.
Functions of TH1 Cells During Antiviral Cellular Immune Responses in the Respiratory Tract
Classic functions of TH1 cells are to support activation and proliferation of the CD8+ CTLs. TH1 cells are the most abundant subset in the respiratory tract after influenza infection. The lung environment during influenza infection favors the TH1 development and/or induction of TH1 migration into the lung. The majority of TH1 cells also produce TNF-α and IL-2, in addition to IFN-γ (139). Establishing a CTL response to eliminate the virus-infected cells requires interactions between three rare cells: viral antigen-presenting DCs, cognate IFN-γ-producing TH1 cells, and cognate CTLs. CD103+ DCs capture the viral antigens in the lung and airways and then migrate into the draining lymph nodes (91). Lymph-node-resident DCs receiving antigens from migrated DCs or migrated DCs themselves are licensed with cognate TH1 cells through CD40–CD40L signaling. The licensed DCs activate the cognate CD8+ T cells to develop and proliferate into functional CTLs (90). IFN-γ produced by TH1 cells is needed to activate the CD8+ T cells. Mature CD8+ CTLs recognize and kill the virus-infected cells, and some cells remain after viral clearance to become resident memory CD8+ T cells, prepared for the next infection with the same pathogen (109,132). Although DCs can memorize licensing information, the chance of interacting with the cognate CD8+ T cells is low. How DCs and T cells overcome this problem is unknown. Virus-specific CTL responses are observed in secondary influenza viral infection; in addition, CTL functions are required for viral clearance in primary influenza infections (125). IFN-γ-producing TH1 cells contribute to activation of primary and secondary antiviral CTLs (120).
Cytotoxic CD4+ T cells are CD4+ T cells with cytotoxic capabilities that depend on the MHC class II, and these cells have been identified from mice to humans (21,36,40,77, 81,127). Cytotoxic CD4+ T cells show TH1-like signatures and may be difficult to distinguish from TH1 cells. In mice infected with lymphocytic choriomeningitis virus (LCMV), cytotoxic CD4+ T cells kill the splenocytes pulsed with LCMV peptides (60). The same phenomenon is observed in mice infected with influenza virus and West Nile virus (12,14). Cytotoxic CD4+ T cells isolated from the lung of mice infected with influenza virus kill the target cells pulsed with viral peptides (121).
Responses and Functions of the Helper T Cells in Lungs Infected with Influenza Virus
Influenza virus infections and CD4+ T cell responses to lung infections are the best studied. Once influenza virus infects the host, the viral replication cycle is extremely fast, ∼6 h. Viral load in the respiratory tract reaches a plateau by 3 days after infection (118). Because virus-specific T cells appear as early as 5 days after infection, the contribution of T cell-mediated immune responses to prevention of initial viral replication is limited. However, T cell-mediated immune responses contribute to influenza virus clearance, preparing for secondary infections. CD8+ T cells have a significant role in virus clearance from the lung. Mice with attenuated CD4+ function can eventually clear the influenza virus; however, CD4+ T cells clearly contribute to CTL activity and antibody responses against the virus (125). DCs are the major antigen-presenting cells (APC) capturing the influenza viral antigens by efferocytosis in the lung. DCs are activated by cytokines and viral-RNA-sensing signaling and then migrate into dLN. Cognate CD4+ cells interact with DCs presenting influenza viral antigenic peptides and differentiating into distinct helper T cell subsets through cytokine signaling.
Infection with influenza virus is a potent IFN-γ-inducing event. IFN-γ-producing TH1 cells dominate the other helper T cell subsets in the respiratory tract after influenza virus infection. Brown et al. elegantly demonstrated development of the TH1 cells in the lung using a virus-specific T cell receptor transgenic mouse model. The adoptive transfer of virus-specific CD4+ T cells into these mice induces the IL-2-producing CD4+ T cells in dLN after infection (15,16). These proliferating IL-2-positive T cells migrate into the lung and become terminal effector TH1 cells producing IFN-γ and TNF-α. The requirement for IFN-γ from TH1 cells for virus control depends on the cellular context. Transfer of whole lung lymphocytes from Ifng-deficient mice can lead to clearance of influenza virus from recipient mice. On the contrary, transfer of purified CD4+ T cells from Ifng-deficient mice does not lead to viral clearance, although cells from wild-type mice are capable of effecting clearance (16).
TFH cells develop efficiently in the spleen and dLN after influenza infections, but they are relatively rare in the NALT and lung (11). iBALT in the lung contains the GCs and requires TFH cells for development. However, iBALT formation is observed during the resolution phase of infection, when antivirus Ig responses have already occurred (88). Thus, the requirement for TFH cells in the lung for the initial protective antiviral Ig responses, especially the IgA response, is not clear. The GC–TFH cells in iBALT are probably required to establish long-lived plasma cells, memory B cells, and the cross-strain reactive B cells (1).
Development of TH2 cells in mice infected with influenza virus has been previously described (5,20); however, influenza infection reportedly inhibits the recruitment for TH2 cells into the airways (130). Helper T cells producing IL-4, one of the signature TH2 cytokines, are induced by DCs activated by viral infection, but not by DCs activated by inactivated vaccines. This suggests that TH2 differentiation requires the highly activated DCs (95). Chronic asthma is a risk factor for severe respiratory symptoms caused by influenza infection (75). Thus, correlations between virus infection and TH2-mediated asthma have been studied. Adoptive transfer of TH2 cells specific against influenza viral antigens induces antigen-dependent lung eosinophilia (72). However, during influenza infection, TH1-derived IFN-γ mediates the neutrophilic dominance over eosinophils attracted by the TH2 cytokines. Whether TH2 cells contribute to influenza-associated asthma exacerbation is not well studied. Acceleration of the IL-33–IL-13 axis by influenza infection induces lung eosinophilia through a mechanism independent of both T and B cells. Thus, innate lymphoid cells are another possible cell type responsible for asthma exacerbation after influenza infection (23).
TH17 cells were discovered as effector T cells responsible for autoimmune diseases (9). IL-23 maintains the TH17 cells in peripheral tissues. TH17 cells produce the signature cytokine IL-17 in addition to IL-21, IL-22, and TNF-α. Contribution of TH17 cells in respiratory viral infections has not been studied extensively.
TH17 cells also are reportedly present in the lungs of influenza-infected mice. IL-17a deficiency in mice reduces lung inflammation caused by influenza infection, although the mice can clear the virus (29). Development of TH17 cells after vaccination with influenza antigens increases lung inflammation and morbidity on challenge with the virus (83). Thus, TH17 cells may be involved in respiratory viral pathogenicity. However, adoptive transfer of TH17 cells can protect the recipients against lethal viral infections (84). This shows the potentially protective function of TH17 cells in some circumstances. TH17 cells in the lung of mice infected with influenza virus are a minor population. Type I IFNs induced by influenza infection diminish IL-23 production, which is required for TH17 cell maintenance (68). IFN-γ and IL-10 induced by influenza infection also inhibit TH17 differentiation (84). Because TH17 cells play a significant role in the defense against bacterial infections in the lung, attenuation of TH17 function may lead to severe bacterial infections after an influenza infection.
Regulatory T cells (Tregs) are categorized into two different subsets: natural Tregs (nTregs) and peripheral Tregs (pTregs). nTregs differentiate in the thymus and regulate immune reactions to self-antigens. pTregs differentiate in the peripheral tissues and regulate immune reactions to both self-antigens and foreign antigens (107). Foxp3, which is the master transcriptional factor for Treg differentiation in mice, is induced through TGF-β signaling (96). Tregs produce IL-10 and TGF-β to repress development and function of the other T cell subsets. Tregs inhibit the conventional T cell proliferation by abrogation of TCR signaling and IL-2 signaling through direct cell–cell contact (110). Transition among the different T cell subsets occurs in some situations due to their plasticity, and this may correlate with disease onset (4,54,80). Tregs accumulate in the lung, airways, and dLN during the acute phase of influenza infection (7,10). Accumulated Tregs inhibit the functions of CD4+ and CD8+ effector T cells. Interestingly, lung Tregs express T-bet, which is a signature transcriptional factor in TH1 cells, but Tregs in the dLN do not. CXCR3 regulated by T-bet augments formation of the Tregs into the lung to prevent the tissue damage caused by excessive CTL activity (45). Viral inhibition effected by Tregs in neonatal mice has also been reported (98). Tregs may induce development of the virus-specific TFH cells by restricting IL-2 availability in the mice infected with influenza virus (71). Memory Tregs accumulate in the lung and dLN during secondary influenza virus infections and efficiently control the virus-specific CD8+ T cell functions (13).
In influenza virus infection, virtually all kinds of the helper T cell subsets develop in the respiratory tract. TH1 cells are the most abundant in the lung and contribute to inhibition of virus replication. The innate immune system–TH2 axis contributes to allergic responses in the respiratory tract during the resolution phase after virus clearance. TH17 and Tregs are relevant to immune responses induced by the influenza virus.
The TH1–TH2 Balance Determines the Pathogenicity of RSV Infections
TH2 cells and TH2 cytokines reportedly contribute to the pathogenesis of RSV infections in humans and mice (44,70,136). In a mouse model of RSV-induced respiratory inflammation, IL-4 depletion decreases lung inflammation (26). Another TH2 cytokine IL-13 is also tightly correlated with RSV-induced lung diseases (78,117). IL-13 inhibition reduces mucus production and airway resistance (122). TH1-mediated immunity induced by immunization with the recombinant bacillus Calmette–Guérin carrying the RSV antigen protected against RSV infection in a mouse model (17,22). Taken together, dominance of the TH2 immunity compared to the TH1 immunity causes severe diseases in the RSV infections.
Mechanisms underlying the TH1–TH2 balance during RSV infection have not been elucidated. NS1 and NS2 RSV proteins directly counteract the type I and type III IFNs, but not type II IFN signaling (116). Munir et al. showed that RSV decreased IFN-γ production in a coculture system comprising DCs and autologous CD4 T cells through the RSV NS1 protein (89). Type I IFN exerts a dual role in the helper T cell differentiation; many reports have shown both stimulatory or inhibitory effects (34,39,51,124). Thus, RSV infection inhibits the TH1 responses and/or augments the TH2 responses by modulating the DC function and through some other unknown mechanisms.
TH17 cells have been found in the lung of RSV-infected mice and humans (8,18). It is not clear if the presence of TH17 cells in RSV-infected patients is beneficial or pathogenic (18); however, in a mouse RSV model, TH17 induces IL-13 and IL-21 and causes mucus production in the respiratory tract (63). Moreover, IL-17 from TH17 cells suppresses the CTL activity that kills RSV-infected cells (19). Thus, TH17 cells may contribute to pathogenicity in RSV-infected patients.
In mouse models, attenuation of Tregs by Foxp3 depletion or anti-CD25 treatment increases migration of the effector T cells into the lung, and inflammatory responses delay recovery (32,38,69). IL-10-deficient mice and mice treated with an IL-10 receptor inhibitor show severe lung inflammation during RSV infections (76). Attenuation of IL-10 in Rag2−/− mice and depletion of CD4+ T cells in the mouse lung after infection indicate that CD4+ T cells are the major source of IL-10 (128). IL-10 producing CD4+Foxp3− type 1 regulatory (Tr1) cells also develop and inhibit inflammation in the lungs of RSV-infected mice (76). A reduction in the frequency of activated Tregs was reported in the peripheral blood of RSV-infected infants (25,67). Thus, Tregs may play a significant role in limiting lung inflammation during RSV infection.
Modulation of the TH1–TH2 balance by RSV infection correlates with respiratory diseases. The mechanisms underlying this balance have been elucidated and will facilitate establishing better treatment regimens.
Helper T Cells in Coronavirus and Rhinovirus Infections
Recently, respiratory coronaviruses, such as those causing severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome, have emerged and resulted in high mortality of infected patients (24). Respiratory coronaviruses cause severe symptoms such as acute respiratory distress syndrome and multiorgan failure. In the acute phase of coronavirus infection, severe leukopenia is seen. Reportedly, CD4+ and CD8+ T cell numbers were reduced in over 80% of SARS coronavirus-infected patients (73,131). The low number of T cells is correlated with symptom severity. SARS coronavirus replicates in epithelial cells but not in T cells (112). The mechanisms underlying T cell depletion by SARS coronavirus are not clear. Dysfunction of APCs and inhibition of DC migration into the lymph nodes may contribute to T cell depletion (135,138). Later in infection, coronavirus-specific CD4+ and CD8+ T cells develop and may contribute to lung pathology by these coronaviruses. Slow T cell expansion is correlated with the severity of disease (135,138). However, in mice infected with mouse hepatitis virus type 1 (MHV-1), which is a group 2 coronavirus, alveolar macrophages induce virus-specific CD4+ and CD8+ T cell responses in the lung, which contribute to MHV-1-induced disease. MHV-1 infection also increases the number of the Foxp3+ Tregs in the lung (49). Further investigations are needed to reveal the T cell suppression mechanisms likely driven by coronavirus infections in humans.
Rhinovirus infection results in common cold-like symptoms and sometimes asthma exacerbation (42). Rhinovirus induces TH2 immunity and eosinophil attraction into the lungs, causing increase of airway resistance. TSLP and IL-33 axis correlates with rhinovirus-induced asthma exacerbation (6,57,59). TSLP, IL-33, and IL-25 are released from epithelial cells through activation of Toll-like receptor signaling in response to rhinovirus infections (2,62,64). Subsequently, TSLP, IL-33, and IL-25 activate DCs, which induce differentiation of the TH2 cells producing IL-4, IL-5, IL-9, and IL-13 (115). These TH2 cytokines attract and activate eosinophils and basophils, cells that effect asthma exacerbation (35,82). The blockage of IL-33 and TSLP signals enforces natural Treg development and reduces lung inflammation in a mouse asthma model (85). Thus, the balance between TH2 and Treg determines lung pathology due to rhinovirus infections.
Conclusions
Effector T cell responses in different respiratory viral infections differ strikingly. Determinants of these responses are largely unknown, but may involve viral accessory proteins as immune modulators, the viral replication speed, differences in activation of the innate immune system, or viral tropism for specific immune cells in addition to the epithelium.
TH1 and TFH cells are the major helper T cell subsets involved in influenza infection. TH1 cells mediate the CTL responses to clear virus-infected cells. TFH cells with high affinity and memory humoral immunity are used to prevent secondary infections by the same viruses. TH17 cells are involved in both lung inflammation and virus clearance during influenza infections. Tregs inhibit excessive immune responses to avoid lung injury. Upregulation of TH2 immunity in RSV infection by largely unknown mechanisms contributes to lung damage due to associated viral virulence. During respiratory coronavirus infections, the helper T cells are significantly diminished by disruption of the antigen-presenting system. Exacerbation of asthma by rhinovirus infection is tightly correlated with the TH2 immunity.
A deeper understanding of the helper T cell development and functions helps to control effector responses that determine viral disease and memory immunity in the respiratory tract.
Footnotes
Acknowledgment
The author thanks Masato Kubo of Tokyo University of Science for valuable suggestions and comments.
Author Disclosure Statement
The author declares no conflicts of interest associated with this article.