
Abstract
Superoxide dismutase 2 (SOD2) is essential in radical scavenging, which balances the intracellular level of reactive oxygen species (ROS). The dysfunction of SOD2 is associated with increasing incidence of various human diseases, including cancer, neuron diseases, and myocardial defects. However, the connections between SOD2-mediated oxidative homeostasis and innate immune response remain unclear. In this study, we report that SOD2 is a crucial regulator of antiviral signaling. Depletion of SOD2 impairs RNA virus-induced type I interferon (IFN) and proinflammatory cytokine production, resulting in enhanced viral replication. Type I IFN production is highly sensitive to cellular level of ROS. SOD2 deficiency-mediated ROS accumulation potently inhibits RIG-I-like receptor (RLR)-induced innate immune responses through the regulation of nuclear factor-kappa B (NF-κB) and interferon regulatory factor-3 activation. These findings uncover a novel role for SOD2 in regulating RLR-mediated antiviral innate immune signaling.
Introduction
T
The cellular stress response is a reaction to defense against internal and external stressful stimuli, including oxidative stress, endoplasmic reticulum stress, heat shock, and DNA damage (7). Increasing evidence suggests a link between cellular stress and innate immune responses (18). Reactive oxygen species (ROS) are by-products of cellular metabolism, including the superoxide anion, hydroxyl radicals, and peroxides. An imbalance between ROS production and ROS elimination is the basis of oxidative stress (1). It has been known that ROS and oxidative stress are involved in the immune responses, such as the activation of inflammasome, regulation of signal transduction, and production of cytokines (24). ROS can also be produced by phagocytes after bacterial infections and exert antimicrobial actions (5). Thus, the components of oxidative stress may have impact on the immune response to infectious pathogens.
Superoxide dismutases (SODs) are crucial antioxidant enzymes that catalyze superoxide radicals into oxygen or less reactive hydrogen peroxide. There are three forms of SODs: cytosolic SOD1, mitochondrial SOD2, and extracellular SOD3 (34). SOD2, also known as manganese-dependent superoxide dismutase (MnSOD), is essential in radical scavenging, which balances the intracellular level of ROS (13). Mitochondria are a major source of ROS production due to electron leakage along the respiratory chain. Loss of SOD2 activity leads to increased oxidative damage in mitochondria and alterations in mitochondrial function (32). SOD2-deficient mice develop mitochondrial disease and die soon after birth (17). In humans, the dysfunction of SOD2 is associated with various diseases, including cancer, neuron diseases, and myocardial defects (3,6). Therefore, SOD2 is critical in the antioxidant defense systems and responsible for oxidative homeostasis in cells.
Although the role of SOD2 in scavenging ROS is well established, it is unknown whether SOD2 is involved in antiviral immunity. In this study, we demonstrate that SOD2 facilitates IFN production and is critical for cellular antiviral responses. In the absence of SOD2, this leads to impaired antiviral cytokine production and increased viral replication in the virus-infected cells. The silence of SOD2 enhanced the intracellular level of ROS, resulting in attenuated RLR-induced type I IFN responses through the regulation of NF-κB and IRF3 activation. Our results describe a previously unknown function for SOD2 in antiviral immunity.
Materials and Methods
Cell culture and reagents
HEK293T and Vero cells were obtained from Type Culture Collection of the Chinese Academy of Science. The J774A.1 cell line was a kind gift from Dr. Guang Yang (Jinan University, China). The 2fTGH-ISRE-Luc cell line was a kind gift from Dr. Fuping You (Peking University, China). The cells were cultured at 37°C under 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). Low molecular weight (LMW) and high molecular weight (HMW)-PolyI:C were from Invivogen. Rotenone and N-acetyl cysteine (NAC) were from Sigma. The antibodies specific to SOD2, IRF3, and phosphor-IRF3 (S386) were purchased from Abways. The antibody specific to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Sungene Biotechnology.
Viruses
Sendai virus (SeV) was from Dr. Bo Zhong (Wuhan University, China). Vesicular stomatitis virus with green fluorescent protein (GFP) (VSV-GFP) provided by Dr. Fuping You (Peking University, China) was passaged once in Vero cells, and viral plaque-forming units (PFUs) were quantified by plaque assay.
shRNA and lentiviral infection
The small hairpin RNAs (shRNAs) that targeted human SOD2 were as follows: shSOD2-1 (5′-GTG GTG GTC ATA TCA ATC ATA-3′), shSOD2-2 (5′-GCA CGC TTA CTA CCT TCA GTA-3′), and scrambled shRNA (control) (5′-CAA CAA GAT GAA GAG CAC CAA-3′). Lentivirus was produced by cotransfection of HEK293T cells with shRNA in the vector pLKO.1 puro (Addgene plasmid No. 8453), pCMV-dR8.2 dvpr (Addgene plasmid No. 8455), and pCMV-VSVG (Addgene plasmid No. 8454). Supernatants containing lentivirus were harvested at 36–48 h after transfection and used to infect the target cells for 24 h. To select infected cells, puromycin (2 μg/mL) was added to the media. Protein knockdown was measured by immunoblot analysis.
Quantitative polymerase chain reaction analysis
Total mRNA was harvested from infected and uninfected cells using TRI Reagent (Sigma), and cDNA was synthesized using Reverse Transcription Reagent Kit (TaKaRa, Japan). Amplification was performed using SYBR Green (Biotools, China) with gene-specific primers in a CFX-96 system (Bio-Rad), and values were normalized to the β-actin gene. Primers for quantitative polymerase chain reactions (qPCRs) were synthesized according to customized sequences as follows: SOD2 (forward: 5′-TAA CGC GCA GAT CAT GCA GCT G-3′, reverse: 5′-AGG CTG AAG AGC GAC CTG AGT T-3′), IFNβ (forward: 5′-CTT GGA TTC CTA CAA AGA AGC AGC-3′, reverse: 5′-TCC TCC TTC TGG AAC TGC TGC A-3′), IL6 (forward: 5′-AGA CAG CCA CTC ACC TCT TCAG-3′, reverse: 5′- TTC TGC CAG TGC CTC TTT GCT G-3′), and β-actin (forward: 5′-AGA GCT ACG AGC TGC CTG AC-3′, reverse: 5′-AGC ACT GTG TTG GCG TAC AG-3′).
Luciferase reporter assays
The pGL3-IFNβ luciferase reporter plasmid was kindly provided by Dr. Fuping You (Peking University, China). The pISRE-Luc and pNFκB-Luc plasmids were purchased from Stratagene. The pRL-TK plasmid was purchased from Promega. HEK293T cells were transfected with the IFNβ luciferase reporter, pRL-TK, or the indicated plasmids for 24 h. Then, cells were stimulated with PolyI:C (1μg/mL) or infected with SeV for 16 h. Luciferase activities were measured with Dual-Luciferase Reporter Assay System (Transgene, China) according to the manufacturer's instructions.
Cytokine enzyme-linked immunosorbent assay measurements and Type I IFN bioassays
Supernatants were harvested and stored at −80°C and IL-6 was quantified by enzyme-linked immunosorbent assays (ELISAs) (Biolegend). Type I IFNs were quantified using the 2fTGH-ISRE-Luc cell line (33). In brief, supernatants from infected and uninfected cells were incubated with 2fTGH-ISRE reporter cells for 6 h. Cells were lysed and used for luciferase quantification. A serial dilution of human IFN-β (PBL Interferon Source) was included as standards.
Immunoblot analysis
Cells were harvested using RIPA III lysis buffer containing protease inhibitor cocktail (Biotechwell, China). Equal amounts of proteins were separated by SDS-PAGE and then transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-rad). Immunoblots were probed with anti-SOD2, anti-IRF3, anti-phospho-IRF3 (S386), and anti-GAPDH, developed using enhanced chemiluminescence (ECL) reagents (Thermo Fisher).
Intracellular and mitochondrial ROS measurements
ROS-sensitive dyes were used to measure intracellular ROS (H2DCF-DA, Yeasen, China) and mitochondrial ROS (MitoSOX Red, Invitrogen). In brief, cells were washed with phosphate-buffered saline (PBS) and then incubated for 30 min at 37°C with H2DCF-DA (10 μM) or MitoSOX Red (5 μM) in serum-free DMEM. The cells were washed with PBS and trypsinized. Fluorescence was analyzed by microplate reader or flow cytometry.
Statistical analysis
Analysis was performed with an unpaired Student's t-test (GraphPad Software). Data are presented as mean ± SEM of three independent experiments. Values of p < 0.05 were considered to be statistically significant.
Results
SOD2 is involved in cellular antiviral responses
To explore the potential role of SOD2 in cellular antiviral responses, we designed small hairpin RNA (shRNA) that targeted two sites of human SOD2 and generated SOD2-silenced HEK293T cells. Endogenous SOD2 was silenced efficiently as quantified by immunoblot analysis (Fig. 1A). We examined the viral replication in SOD2-silenced cells infected with VSV-expressing GFP (VSV-GFP). The percent of VSV-GFP-positive cells was increased in SOD2-silenced cells after VSV infection for 12 h (Fig. 1B and Supplementary Fig. S1; Supplementary Data are available online at
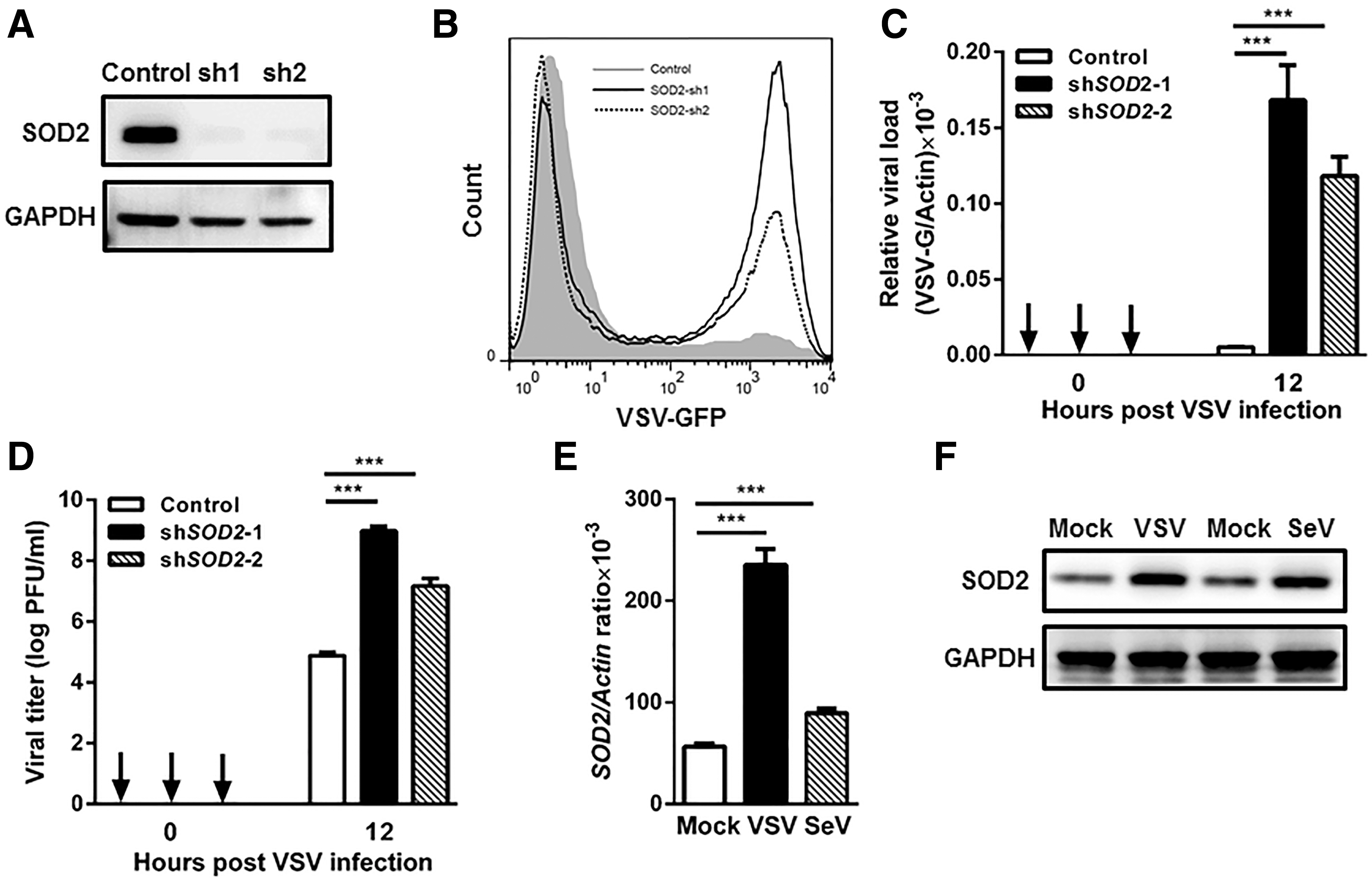
SOD2 is involved in cellular antiviral response.
We next examined the expression pattern of SOD2 upon viral infection. The mRNA and protein expression levels of SOD2 were measured by qPCR and immunoblot in J774A.1 murine macrophages infected with RNA virus. After 12 h of infection, the mRNA level of SOD2 was increased by 4.2-fold in VSV-infected cells and 1.6-fold in SeV-infected cells (Fig. 1E). Immunoblot further confirmed the upregulation of SOD2 protein expression after VSV and SeV infection (Fig. 1F). Together, these data suggest that SOD2 is involved in cellular antiviral responses.
SOD2 regulates RNA virus-induced cytokine production
Induction of type I IFN is a key feature of antiviral innate immunity (8). We assessed the effect of SOD2 on type I IFN response using luciferase reporters. Silencing of SOD2 expression impaired virus-induced IFNβ and ISRE reporter luciferase activity (Fig. 2A, B). We further quantified mRNA expression levels of IFNβ in SOD2-silenced cells by qPCR. Notably, the depletion of endogenous SOD2 significantly reduced the transcription level of IFNβ after VSV and SeV infection (Fig. 2C). In parallel, the decrease in IFN-β production from SOD2-silenced cells was also significant at the protein level (p < 0.05) (Fig. 2D). We also quantified mRNA expression levels of CCL5 and CXCL10 in SOD2-silenced cells by qPCR. The results suggest that silence of SOD2 impaired the production of CCL5 and CXCL10 after VSV and SeV infection (Supplementary Fig. S2). These results suggest that SOD2 may contribute to type I IFN production during virus infection and, consequently, inhibit virus replication.
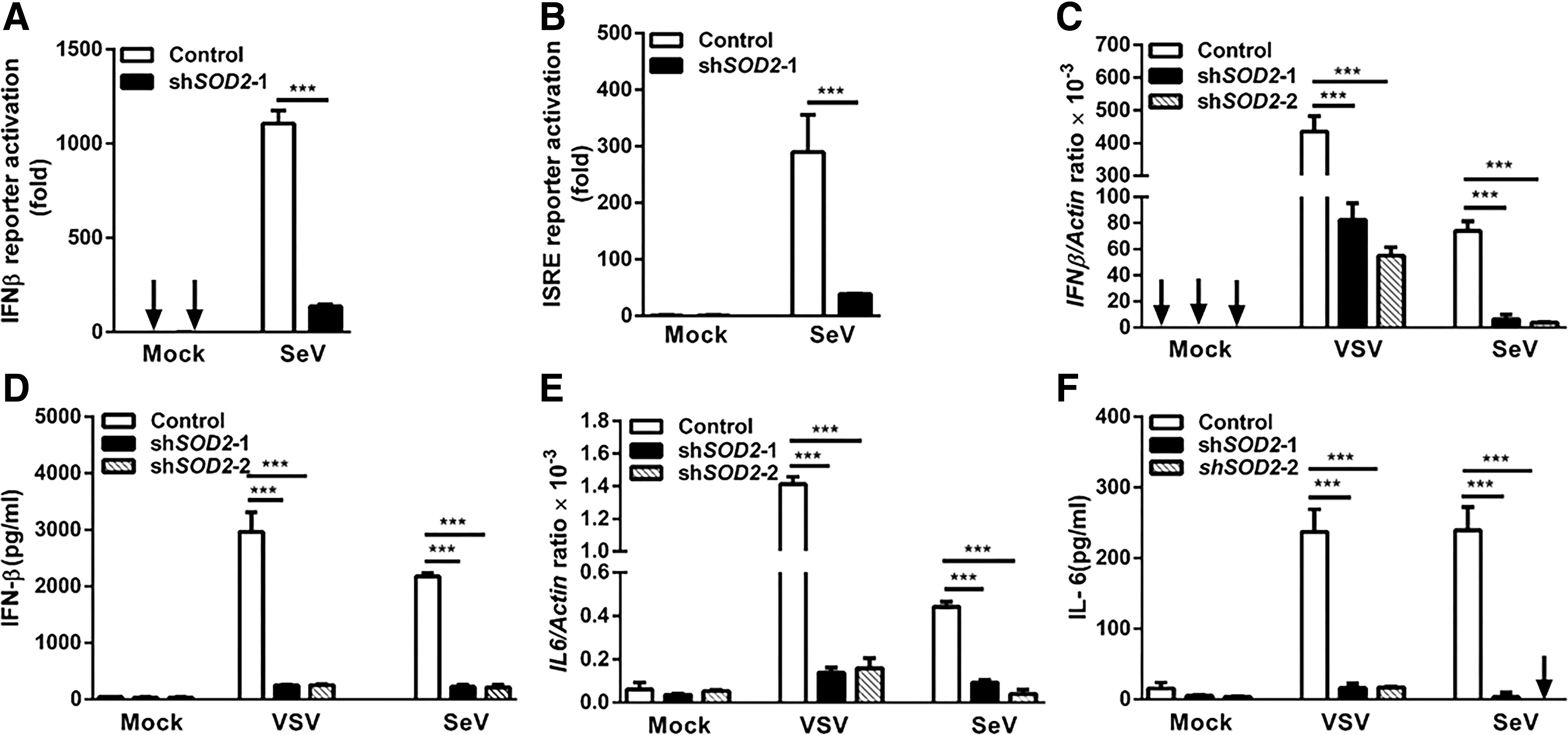
SOD2 regulates RNA virus-induced cytokine production.
In addition to establishing an antiviral state through type I IFN production, a successful antiviral immune response includes the production of proinflammatory cytokines, such as IL-6 (9). We next examined the expression of IL-6 in SOD2-silenced cells. In agreement with the IFNβ findings, these cells had lower mRNA expression level of IL6 after infection with VSV and SeV (Fig. 2E). The protein secretion of IL-6 was also decreased in SOD2-silenced cells (Fig. 2F).Taken together, these findings indicated that SOD2 modulates antiviral immune responses through regulation of cytokine production.
Loss of SOD2 impairs antiviral signaling by producing more ROS
Previous studies have shown that SOD2 proceeds to scavenge ROS in mitochondria (13). Thus, we next examined whether the silence of SOD2 led to the increased level of ROS in HEK293T cells. Through the use of ROS-sensitive dyes, we measured the levels of mitochondrial ROS (staining with MitoSOX Red) and cellular ROS (staining with H2DCF-DA) in SOD2-silenced cells. The mean fluorescence intensity of MitoSOX Red was higher in SOD2-silenced cells, indicating the higher level of mitochondrial ROS in those cells (Fig. 3A). Furthermore, the cellular ROS level was increased by 2-fold in SOD2-silenced cells compared with control cells (Fig. 3B).

Loss of SOD2 impairs antiviral signaling by producing more ROS.
We next asked whether increased production of ROS led to the decreased level of IFN-β production in response to viral infection. Rotenone was used to increase mitochondrial ROS in HEK293T cells. As expected, treatment with rotenone at concentrations that increase mitochondrial ROS resulted in the inhibition of IFNβ mRNA expression after VSV and SeV infection (Fig. 3C). In agreement with these findings, the decrease in IFN-β production from rotenone-treated cells was also significant at the protein level (p < 0.01) (Fig. 3D), while treatment with an antioxidant NAC enhanced the IFN-β production induced by virus infection (Fig. 3E, F). All the results indicate that type I IFN production is highly sensitive to mitochondrial and cellular levels of ROS, and SOD2 regulates antiviral signaling through modulation of the ROS level.
SOD2 deficiency impairs innate immune response through the RLR signaling pathway
The RLR family members RIG-I and MDA5 can recognize viral RNA, then initiate the activation of signaling pathways that lead to production of type I IFN and inflammatory cytokines (16). We examined whether SOD2 regulated RNA virus-induced innate immune responses through the RIG-I/MDA5 pathway. By transfection of synthetic viral mimic PolyI:C (specific agonists of RLR pathway), we evaluated the effect of SOD2 on RLR-dependent IFNβ promoter activity. We found that knockdown of SOD2 attenuated the activation of IFNβ promoter in HEK293T cells transfected with both low-molecular-weight PolyI:C (RIG-I ligand) and high-molecular-weight PolyI:C (MDA5 ligand) (Fig. 4A). In agreement with this finding, overexpression of SOD2 facilitates IFNβ reporter luciferase activity after SeV infection or LMW-PolyI:C transfection (Supplementary Fig. S3). In addition, SeV-induced IFNβ activation was also impaired in ROS inducer rotenone-treated cells (Fig. 4B).These data suggest that RLR signaling is impaired by increased levels of ROS in SOD2 knockdown cells.
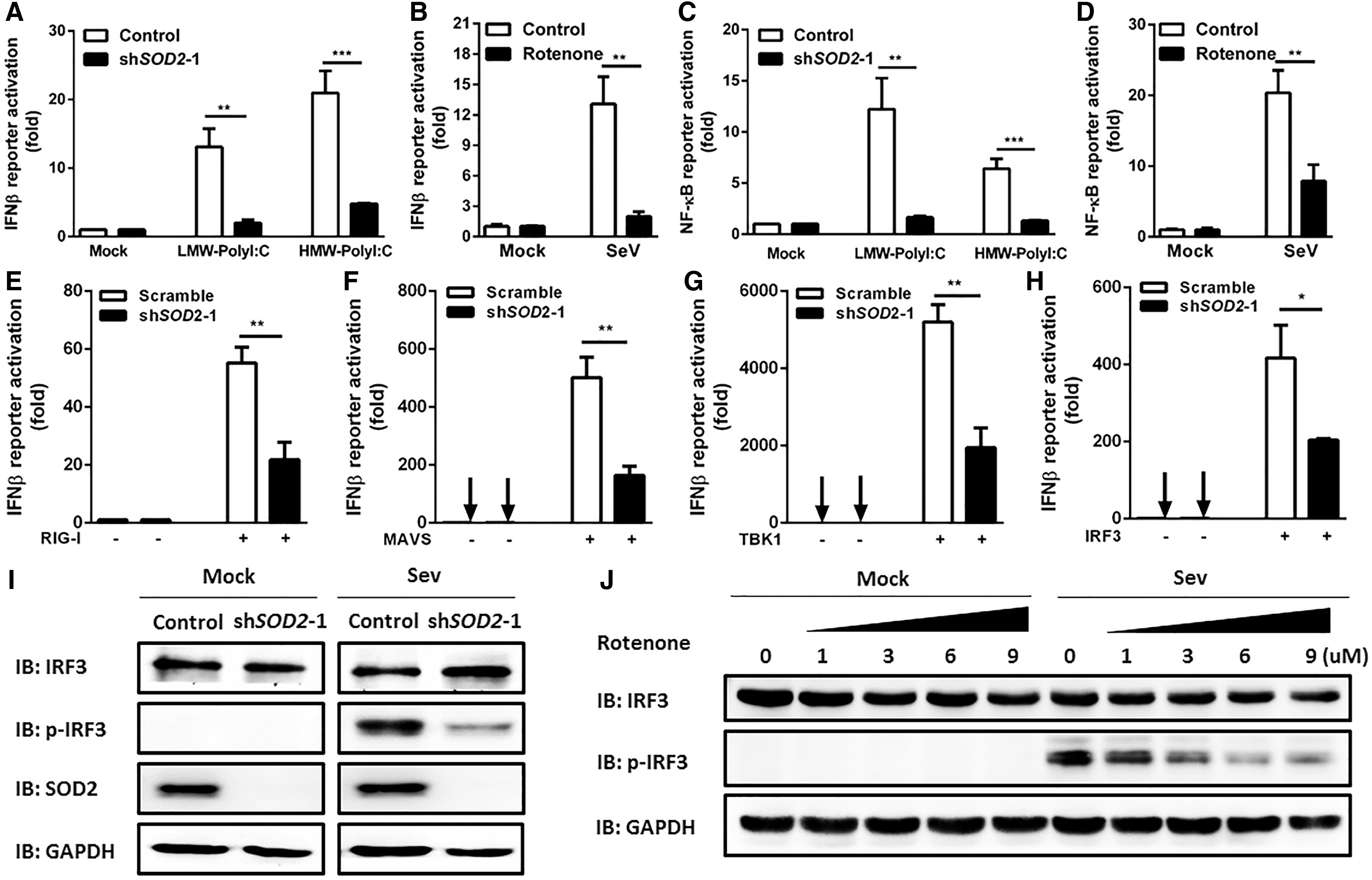
SOD2 deficiency impairs activation of IRF3 and NFκB signaling.
Nuclear factor-κB (NF-κB) is the downstream molecule of RLR pathway and contributes to the production of IFN-β or proinflammatory cytokines (15). We next assessed the impact of SOD2 on RLR signaling-induced NF-κB promoter activation. Results demonstrate that silencing of SOD2 led to reduced activation of NF-κB promoter by transfection of PolyI:C (Fig. 4C). Furthermore, the relative activity of NF-κB was decreased by 2.7-fold in rotenone-treated cells after SeV infection (Fig. 4D).
To identify the potential target of SOD2 in RLR signaling, we overexpressed RIG-I, mitochondrial antiviral signaling (MAVS), TBK1, and IRF3 in control and SOD2-silenced cells and detected the IFNβ reporter luciferase activity. We found that the silenced SOD2 impaired the RIG-I, MAVS, TBK1, and IRF3-induced IFNβ promoter activation (Fig. 4E–H). The IRFs have been recognized as key regulators of type I IFN gene expression. Activation of RLR signaling induces the phosphorylation of IRF3, then induces the transcription of IFNβ (15). We further observed less phosphorylation of IRF3 at Ser386 in SOD2 knockdown cells after SeV infection (Fig. 4I). We next asked whether ROS accumulation by SOD2 silencing impaired the activation of IRF3. Consistent with the above results, phosphorylation of IRF3 was significantly reduced in HEK293T cells pretreated with rotenone in a dose-dependent manner (Fig. 4J). Together, these results indicate that SOD2 plays an important role in RLR signaling-induced IFN-β production through NF-kB and IRF3 pathway.
Discussion
SOD2 is predominantly known as an antioxidant enzyme which participates in multiple biological processes. Loss of SOD2 activity contributes to the increasing incidence of various human diseases (6). In addition, significant evidence links SOD2 alterations and age-related phenotypes. SOD2 overexpression extends the life span of Drosophila. Age-related reduced expression of SOD2 was observed in humans (25,27). Mice with heterozygous deficiency of SOD2 have a skin immune system with features of inflamm-aging (23). ROS are linked with aging. Human studies of age-related defects in antiviral response are associated with oxidative stress (19). Previous studies have shown that ROS are induced after virus infection, such as hepatitis B virus, hepatitis C virus, and influenza A virus infection (11,12,21). Thus, ROS may play an important role in modulating antiviral immunity. We are tempted to speculate that dysfunction of antioxidant enzyme SOD2, which leads to ROS accumulation during aging, may contribute to increased susceptibility to infection in elderly. However, the involvement of SOD2 in the antiviral immune response has not yet been reported. In this study, we investigate whether SOD2 is required for RNA virus-induced host antiviral response. Silencing of SOD2 by RNA interference impairs virus-induced IFNβ promoter activation and IFN-β protein secretion. SOD2 protein expression is induced following viral infection. These findings suggest that SOD2 is involved in cellular antiviral responses. This was further strengthened by our observations that knockdown of SOD2 enhances viral replication. Thus, our study has revealed a novel role of SOD2 in antiviral innate immunity.
Oxidative stress results from an imbalance between oxidants and antioxidants, regulating PRR signaling and immune response to infection. Induction of type I IFN is a key feature of antiviral innate immunity and highly regulated process. The initial production of IFN-α/β is mediated mainly in an IRF3-dependent manner after viral infection. In the late phase, the secreted type I IFN is recognized by IFNAR and activates the signaling cascade of JAK-STAT pathway, including amplification of IFN responses through a positive feedback loop (8). Oxidative stress can influence antiviral immune responses through regulation of the JAK-STAT pathway (4). The receptor tyrosine kinase AXL is induced in response to oxidative stress and limits IFN-β production through hijack IFNAR-STAT1 cassette (19,22). Similarly, the components of the antioxidant system have impact on the production of type I IFN. The antioxidant molecule myeloid HO-1 is required for activation of IRF3 after pathogen infection. HO-1-deficient macrophages showed reduced expression of IFN-β (29). Consistent with these findings, our data show less phosphorylation of IRF3 in SOD2 knockdown cells after viral infection. Of note, ROS accumulation reduces the RNA virus-induced activation of NF-κB and IRF3. These data suggest a further role for oxidative stress in inhibition of RLR signaling-induced IFN-β production through NF-kB and IRF3 pathway.
Mitochondria-localized SOD2 is critical to balance the cellular level of ROS (13). Our findings reveal the connections between SOD2-mediated oxidative homeostasis and innate immune response. Mitochondria have emerged as signaling organelles that contribute to innate host response (30). Upon viral infection, RIG-I and MDA5 translocate to the mitochondria and interact with MAVS adapter. MAVS then activates TBK1-IKKe complexes, which in turn activate the NF-kB and IRF3 (16). Several mitochondrial proteins, such as MUL1 and Tom70, have been reported to interact with MAVS components and regulate IFN production (10,14). However, the direct interaction between SOD2 and MAVS was not observed in our study (data not shown). Mitochondrial dynamics is another layer of complexity to govern antiviral signaling. Mitochondrial fusion protein Mfn1 regulates mitochondrial dynamics to facilitate mitochondrion–endoplasmic reticulum interactions during viral infection, enhancing the activation of RLR signaling (2). Previous report suggests that cellular and mitochondrial redox homeostasis is linked to mitochondrial dynamics (31). Our results suggest that increased ROS level due to SOD2 silencing may influence the mitochondrial homeostasis and then modulate IFN signaling.
In summary, SOD2 facilitates the antiviral innate immune response by maintaining oxidative homeostasis. Our findings describe a critical role of oxidative stress in regulation of immune signaling. An improved understanding of the underlying mechanisms of this regulation will provide potential therapeutic approach in viral infection.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81370464, 81671393), Doctoral Fund of Ministry of Education of China (No. 20130071120031), Shanghai Hospital Development Center (No. SHDC12014221), and Shanghai Municipal Commission of Health and Family Planning, Key Developing Disciplines (2015ZB0501).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.