
Abstract
The extensive hypervariability of human immunodeficiency virus type-1 (HIV-1) populations represents a major barrier against the success of currently available antiretroviral therapy. Moreover, it is still the most important obstacle that faces the development of an effective preventive vaccine against this infectious virus. Indeed, several factors can drive such hypervariability within and between HIV-1 patients. These factors include: first, the very low fidelity nature of HIV-1 reverse transcriptase; second, the extremely high HIV-1 replication rate; and third, the high genomic recombination rate that the virus has. All these factors together with the APOBEC3 proteins family and the immune and antiviral drugs pressures drive the extensive hypervariability of HIV-1 populations. Studying these factors and the mechanisms that drive such hypervariability will provide valuable insights that may guide the development of effective therapeutic and preventive strategies against HIV-1 infection in the near future. To this end, in this review, we summarized recent advances in this area of HIV-1 research.
Introduction
H
By the end of 2015, ∼78 million individuals have become infected with HIV since the start of HIV/AIDS epidemic in the early 1980s, and more than 36 million individuals are currently living with HIV across the whole world according to the reports of UNAIDS (
Perhaps the clearest indication of the extensive hypervariability of HIV-1 populations is the extreme variation in viral genome sequences even within a single HIV-1-infected individual few years post-infection (95), taking into consideration that most HIV-1 infections result from a single virus (transmitted/founder virus) (91,95). The variation of viral genome sequences is much greater between HIV-1-infected patients. One of the selective benefits of the extensive diversity of HIV-1 genome sequences is to efficiently escape the antiviral attacks of the immune system and synthetic antiviral agents (8,202), ensuring HIV-1 persistence in infected individuals.
To control HIV-1 infection, it is essential to create new effective therapeutic and preventive strategies against this grim infectious virus, which, however, cannot be achieved without overcoming the HIV-1 hypervariability problem. One way that could help achieve this goal is to study the mechanisms and factors that drive the extensive HIV-1 hypervariability. To this end, in this review, we aim at summarizing recent advances in this area of HIV-1 research and at suggesting potential strategic solutions to compact HIV-1 hypervariability.
Overview of HIV-1 Structure and Life Cycle
Before starting discussions, it is more convenient to introduce the reader into the general HIV-1 structure and life cycle to clearly understand some points mentioned in the subsequent discussions. In brief, each mature replication-competent HIV-1 particle has two copies of single-stranded (plus-sense) RNA; each strand encodes three major genes that are usually found in all retroviruses, including the pol, gag, and env genes. HIV-1 has also six accessory genes (nef, rev, tat, vif, vpr, and vpu).
The pol gene of HIV-1 encodes for three enzymes that are essential for HIV-1 replication and maturation; these enzymes are the reverse transcriptase (RT), integrase, and protease. HIV-1 gag gene encodes for core structural proteins, including the matrix (P17), two spacer peptides (SP1 and SP2), capsid (P24), gag-polypeptide associated protein (P6), and nucleocapsid (P7). However, HIV-1 env gene encodes for the viral glycoproteins (trans-membrane glycoprotein 41 “gp41” and surface glycoprotein 120 “gp120”) that are associated with its envelope (plasma membrane derived from host cells). Noteworthy, certain viral genes evolve rapidly (e.g., env is the fastest evolving HIV-1 gene) (69), whereas others are more conserved due to their functions (e.g., HIV-1 gag gene).
HIV-1 life cycle starts when a replication-competent HIV-1 encounters a suitable permissive target cell (mainly CD4+ T cells and macrophages) expressing receptors (CD4 as a primary receptor and CCR5 and/or CXCR4 as coreceptors) that are required for binding to the viral surface gp120 (196). This binding results in conformational changes in the structure of gp120 that trigger the unfolding of the viral trans-membrane gp41. In turn, this facilitates the fusion of the viral envelope with the target cell membrane. Next, the viral capsid is released into the cytoplasm of the target cell, where it will be targeted for degradation, releasing two copies of HIV-1 RNA strands with their associated molecules and the three encapsidated replication enzymes (26). Shortly thereafter, HIV-1 RT starts the reverse transcription of viral RNA into DNA (86).
Nonetheless, this scenario is challenged by recent studies that indicated that the reverse transcription occurs within the viral capsid during cytoplasmic transit (Fig. 1), as a strategy to protect viral DNA from the cytosolic DNA sensors and to facilitate the infection of non-dividing cells (“reviewed in 26”) (89). HIV-1 RT uses genomic viral RNA (plus strand) and host transfer RNA to start the synthesis of complementary DNA (negative viral DNA strand), creating an RNA-DNA hybrid. The RNA strand of this hybrid is now subject to cleavage by HIV-1 RT ribonuclease H (RNase H) except two small RNA segments (purine-rich sequences), which, in turn, serve as primers to the synthesis of the second DNA strand. Eventually, these RNA segments are replaced by DNA segments, resulting in a double-stranded viral DNA (dsDNA).

Scenarios of HIV-1 reverse transcription inside and outside viral capsid, and possible mechanisms of dNTPs and divalent cations incorporation into the reverse transcription complex. Literature shows that HIV-1 reverse transcription could occur either inside (case number 1 “the upper left corner”; the early reverse transcription events occur before the viral capsid is targeted for degradation, or the reverse transcription is completely finished before the viral capsid is targeted for degradation as seen in case number 2 “the lower left corner”) or outside (case number 3 “the upper right corner”) the viral capsid in the cytoplasm of an HIV-1-infected cell. In case numbers 1 and 2, the incorporation of dNTPs into the reverse transcription complex through the positively charged pores explains how the reverse transcription process takes place inside the viral capsid as recently published in Nature (89). Similarly, we propose that divalent cations could also be incorporated into the reverse transcription complex through certain pores in the viral capsid, which are not yet defined. In case number 3, the incorporation of both dNTPs and divalent cations is conceivable because HIV-1 reverse transcription takes place within the cytoplasm of an infected cell after degradation of the viral capsid. Finally, the incorporation of dNTPs and divalent cations into the viral capsid during the assembly/budding process could also provide another possible mechanism to explain HIV-1 reverse transcription inside the viral capsid, as seen in case number 4 (the lower right corner). dNTPs, deoyribonucleoside triphosphates; HIV-1, human immunodeficiency virus type-1.
Once the reverse transcription event is finished, the viral integrase imports the viral dsDNA into the nucleus, where it will be integrated into the host DNA (45). Then, the virus exploits the host machineries to transcribe the integrated viral DNA (proviral DNA) into RNA and then translate this RNA into viral proteins (polyproteins). As a result, structural and non-structural viral proteins, in addition to two viral RNA strands, are trafficked and packaged beneath the cell membrane, where the viral assembly/budding process starts, end up by releasing immature viral particles (176). Finally, the viral protease becomes activated and continues the maturation process by cleaving the polyproteins into functional domains (176). The resulting mature HIV-1 particles become capable of infecting new target cells, continuing their replication cycle.
Factors That Drive HIV-1 Hypervariability
The low fidelity nature of HIV-1 RT
According to the general outline of HIV-1 life cycle, the extensive HIV-1 heterogeneity can be related to the genetic mutations that occur during two steps in the viral life cycle: (1) the reverse transcription and (2) the proviral DNA transcription. Similar to other viruses, HIV-1 exploits host machineries to transcribe the proviral DNA, and it then translates the viral RNA transcripts to express its own proteins. Host RNA and DNA polymerases are among these machineries (203). In fact, host DNA polymerase plays a minor role in driving HIV-1 hypervariability due to, at least, two reasons: First, the fidelity of host DNA polymerase is considerably higher than that of RNA polymerase (i.e., RNA polymerase II) and HIV-1 RT (103,191). Second, most HIV-1-infected cells die before they can divide, and, thus, mainly the RNA polymerase II and HIV-1 RT contribute to the errors made during HIV-1 replication.
Even though that RNA polymerase II increases the viral variability through the generation of frame-shift mutations, a common type of mutations that occur during HIV-1 replication, HIV-1 RT is considered the master of this process with more than 97% of the generated HIV-1 frame-shift mutations (203). HIV-1 reverse transcription is an obligatory step in the HIV-1 life cycle (36), since HIV-1s that fail to initiate and/or continue the reverse transcription process fail to continue their life cycles, meaning that their life cycles are restricted at the reverse transcription step. This explains why HIV-1 RT has long been predisposed as a major target for antiretroviral drugs (51). The reverse transcription of the HIV-1 RNA is a very complex process, in which multiple viral and host proteins and factors are involved.
HIV-1 RT is the cornerstone protein in this process, which is a heterodimer enzyme that has two subunits, named p66 and p51 according to their molecular weight (86). The p66 subunit performs both the polymerase and RNase H activities (86), whereas the smaller subunit, the p51, is believed to provide structural support. Studies that uncovered the crystal structure of HIV-1 RT have greatly improved the understanding of mechanisms of HIV-1 dsDNA synthesis from its RNA genome by determining the sites of polymerization and where the incoming deoyribonucleoside triphosphates (dNTPs) are added to the newly synthesized DNA strand (116). These studies have shown that the polymerase domain of HIV-1 RT has “fingers,” “thumb,” and “palm”-like structures, and that is why it is usually compared with a hand. The palm of HIV-1 RT is the active site where the end of a primer is added. Three aspartate residues “negatively charged” (D110, D185, and D186) are known to be involved in the interaction with magnesium ion (Mg2+) associated with the incoming dNTP (93,143).
Noteworthy, specific amino acid residues within the polymerase domain of HIV-1 RT are involved in the primer binding (M184 residue), template binding (E89 residue), and interaction with the incoming dNTPs (K65, L74, V148, and Q151 residues) (116). Importantly, unlike the polymerases of many eukaryotes and viruses, HIV-1 RT has no intrinsic 3′-to-5′ proofreading (exonucleolytic) activity (very low fidelity), making it unable to correct errors that occur during the reverse transcription process (159). Moreover, HIV-1 RT is very efficient at extending the terminus misinserted nucleotides to the newly synthesized DNA strand (13,150). Hence, the low fidelity nature of HIV-1 RT, in part, explains the high genetic hypervariability of HIV-1 populations.
Of note, the fidelity of HIV-1 RT is well recognized to be influenced by different factors, including, but not limited to, certain mutations within the HIV-1 RT itself, divalent cations, and cellular availability of dNTPs (briefly overviewed in Factors That Affect HIV-1 RT Fidelity).
Factors That Affect HIV-1 RT Fidelity
Mutations within HIV-1 RT
The fidelity of HIV-1 RT can be affected by certain mutations within HIV-1 RT itself. For instance, substitution mutations of the K65 residue that present in the p66 subunit can affect the fidelity of HIV-1 RT, since the K65 residue is known to be involved in the binding to and stabilizing of the incoming dNTP by the formation of hydrogen bond with the γ-phosphate of the incoming dNTP.
Garforth et al. have used pre-steady state kinetics to study the impact of K65 in mismatch extension fidelity (66). Their results have indicated that K65 substitution with alanine “A” and arginine “R” led to an enhancement in the HIV-1 RT fidelity via reducing the mismatch extension efficiency (by reducing the binding for the incoming dNTPs) (66). Further, in vitro and in vivo studies have shown that K65R mutation in the RT of both simian immunodeficiency virus (SIV) and HIV-1 can reduce their replication capacities (fitness cost) and enhance the fidelity of RT of SIV and HIV-1 (66,117,192). Intriguingly, although such mutations can enhance the fidelity of HIV-1 RT, they confer cross-resistance to most available nucleoside RT inhibitors (NRTIs), through allowing HIV-1 RT to discriminate against NRTIs analogs, while retaining its capacity to incorporate normal dNTPs (68).
In another instance, Q151N mutation (Q151 residue is known to be involved in the interaction with the incoming dNTP via binding to the 3′-OH on its sugar moiety) has been shown to increase the fidelity of HIV-1 RT by decreasing the stability of incorrect dNTP binding to the Q151N mutant as well as the base-pairing between the incorrect dNTP and the template nucleotide (194). Unlike Q151N mutation, Q151M mutation has the same fidelity as the wild-type HIV-1 RT (94); nonetheless, both mutations confer resistance to different NRTIs (64,188).
Mutation of the highly conserved R72 residue across retroviral polymerases into alanine (R72A) has also been demonstrated to significantly increase the fidelity of HIV-1 RT by enhancing the selectivity of correct dNTPs and decreasing the mismatched primer termini extension (110). In addition, the mutation of R448 residue (which is located in the RNase H domain of HIV-1 RT and interacts with RNA template near the active site of RNase H) has also been shown to affect the enzyme polymerization activity and increase its fidelity by perturbing the interaction with the RNA template (175). On the other hand, other mutations can reduce the fidelity of HIV-1 RT. For example, the substitution mutation of M184 residue by alanine (M184A), but not valine (M184V), has been shown to result in a higher error-prone enzyme (reduced the fidelity of HIV-1 RT) (140).
Indeed, there are many examples of substitution mutations within HIV-1 RT that could also modulate the fidelity of HIV-1 RT and confer cross-resistance to different NRTIs or non-NRTIs such as L228I, Y232H, K103N, and Y181C mutations (68,105,129,204). Other types of mutations such as deletion or insertion mutations, which occur, for example, within the β3–β4 or β7–β8 loops of HIV-1 RT, could also impact the fidelity of HIV-1 RT (67,135,163,184). It is, thus, worthy to know that the fidelity of HIV-1 RT is affected by certain mutations within HIV-1 RT, and such mutations could have fitness costs. Therefore, studying the resistance mechanisms of such mutations can aid in developing and designing new effective therapeutic and preventive strategies against the resistant HIV-1 mutants (105).
Divalent cations
The intracellular divalent cations are essential cofactors for activities of both polymerase and RNase H of HIV-1 RT (2,46,62,72,177). At least three to four binding sites for divalent cations are present within the HIV-1 RT (two binding sites in the active site of polymerase and one to two in the active site of RNase H).
Animal cells contain several divalent cations such as: Mg2+, manganese (Mn2+), copper (Cu2+), cobalt (Co2+), and zinc (Zn2+), among others. Notably, Mg2+ is the most abundant intracellular divalent cation, and it is well known to act as a physiological cofactor for a wide array of enzymes (3,31); it is, thus, not surprising to know that Mg2+ functions as a cofactor for both activities of HIV-1 RT under physiological conditions (2). One in vitro study has shown that the fidelity of HIV-1 RT highly depends on the intracellular concentration of Mg2+ and dNTPs availability (31). Interestingly, the lower intracellular Mg2+ concentration has been shown to be associated with the higher HIV-1 RT fidelity (31), but the exact explanation of this observation is still unknown, thus leaving this question up for future investigations.
However, other in vitro studies have demonstrated that several divalent cations, including the early mentioned ones, can be used by HIV-1 RT as alternatives to Mg2+ and also can manipulate the fidelity of HIV-1 RT (2,19,62,63,178). But since they are kept at very low concentrations by the cellular regulatory mechanisms, it was believed that they do not play a considerable role in both activities of HIV-1 RT (2).
To determine whether these assumptions are true or not, Achuthan and DeStefano have investigated the impact of three different divalent cations (i.e., Zn2+, Co2+, and Mn2+) on the fidelity of HIV-1 RT by using both the optimal and suboptimal concentrations required for the activity of HIV-1 RT (2). Interestingly, they have shown that Co2+ and Mn2+ were able to decrease the HIV-1 RT fidelity at high concentrations, but these effects were almost alleviated when optimal concentrations were used, indicating that the fidelity of HIV-1 RT in the case of Co2+ and Mn2+ is concentration dependent. Remarkably, at optimal concentrations, Zn2+ was able to increase the fidelity of HIV-1 RT two- to threefold when compared with Mg2+.
Other studies have shown that the substitution of Mg2+ by Mn2+ increases misincorporation of dNTPs, alters nucleotide specificity, and increases mutation frequencies (30,189). Interestingly, Cases-Gonzalez et al. (30) have shown that in the presence of Mn2+, mispair extension efficiency was significantly higher (about two orders of magnitude) than in the presence of Mg2+. Taken together, these data indicate that divalent cations significantly influence HIV-1 RT fidelity.
There are different possible mechanisms by which divalent cations can affect the fidelity of HIV-1 RT. For example, it has been supposed that Zn2+ supports a different geometry at the active site of HIV-1 RT than that supported by Mg2+, which may result in a decreased rate of misaligned substrates, thus enhancing the fidelity of HIV-1 RT (2). It has also been shown that Zn2+ promotes the formation of a highly stable and slowly progressing RT-complex that minimizes the misinsertion or misalignment of substrates, which reflects the ability of Zn2+ to affect the fidelity of HIV-1 RT (2,62). Intriguingly, Zn2+ was able to enhance the fidelity of HIV-1 RT two- to threefold when compared with Mg2+ (the physiological cation) (62). Of note, under certain conditions, Zn2+ has inhibitory effects; the Zn2+ concentrations used in this study that was capable of inhibiting HIV-RT were two to three orders of magnitude greater than its physiological concentrations in cells.
Although these data indicate that Zn2+ can affect the fidelity of HIV-1 RT, whether high Zn2+ concentrations have beneficial or detrimental effects on the fidelity of HIV-1 RT in vivo remains to be determined. Hence, these studies have suggested caution when considering the use of supplements or natural minerals for HIV-1 patients, especially those containing Zn2+, to avoid unexpected adverse reactions (2,62). However, additional in vitro and in vivo studies are required to address these assumptions and to uncover the mechanisms by which Zn2+ and other divalent cations can affect the fidelity of HIV-1 RT in vivo. This, in turn, may provide new potential therapeutic targets to inhibit the activities of HIV-1 RT, thereby inhibiting HIV-1 replication.
On the other hand, it is essential to know that divalent cations bind to specific amino acid residues within the HIV-1 RT. Therefore, certain mutations in HIV-1 RT could affect its fidelity, taking into consideration that divalent cations (such as Mn2+) can affect nucleotide substrate specificity in different polymerases, including HIV-1 RT (186). For example, D186 residue is a part of the metal ion binding site. D186H mutation has been shown to affect the DNA polymerase and RNA replicase activities of HIV-1 RT (186). In another example, substitution mutation of E89 residue (E89G), which is located within the p66 subunit, has also been implicated in the alteration of Mg2+ performance, thus affecting the polymerization activity of HIV-1 RT (97), which, in turn, affects the fidelity of HIV-1 RT. Other studies have demonstrated that substitution mutation of E478 residue (E478Q), which is located within the p66 subunit and participates in metal ion binding, has completely inactivated HIV-1 RNase H activity in the presence of Mn2+, but not Mg2+, indicating that Mn2+ is essential for RNase H activity of HIV-1 RT (37).
Finally, it is worthy to know that there are two scenarios to describe the reverse transcription process, inside- and outside-viral capsid scenarios, as introduced in the viral life cycle (26). Accordingly, the incorporation of divalent cations into the RT-complex is conceivable according to the first scenario. Although the mechanisms of divalent cations incorporation into the RT-complex within the viral capsid are still undetermined (Fig. 1), additional investigations are required to determine how the virus imports divalent cations into the viral capsid. As a result, this may uncover new potential therapeutic targets to inhibit or, at least, limit HIV-1 reverse transcription by blocking the incorporation of divalent cations into the viral capsid according to the second scenario.
Availability of dNTPs
The intracellular level of dNTPs is another critical factor that affects the fidelity of HIV-1 RT, since the cellular levels of dNTPs were shown to be directly associated with the frequency of HIV-1 mutations. In other words, the intracellular dNTPs are the substrates for HIV-1 DNA synthesis catalyzed by HIV-1 RT; therefore, their levels are directly associated with the HIV-1 replication rate, which, in turn, is directly associated with the mutation rate observed in the viral reverse transcripts during the replication events (Fig. 2) (96,127). The very rapid replication rate is a characteristic feature of HIV-1 that explains its extensive genetic variability, especially when compared with the genetic variability of other viruses that have similar error rates but have lower replication rates (86).
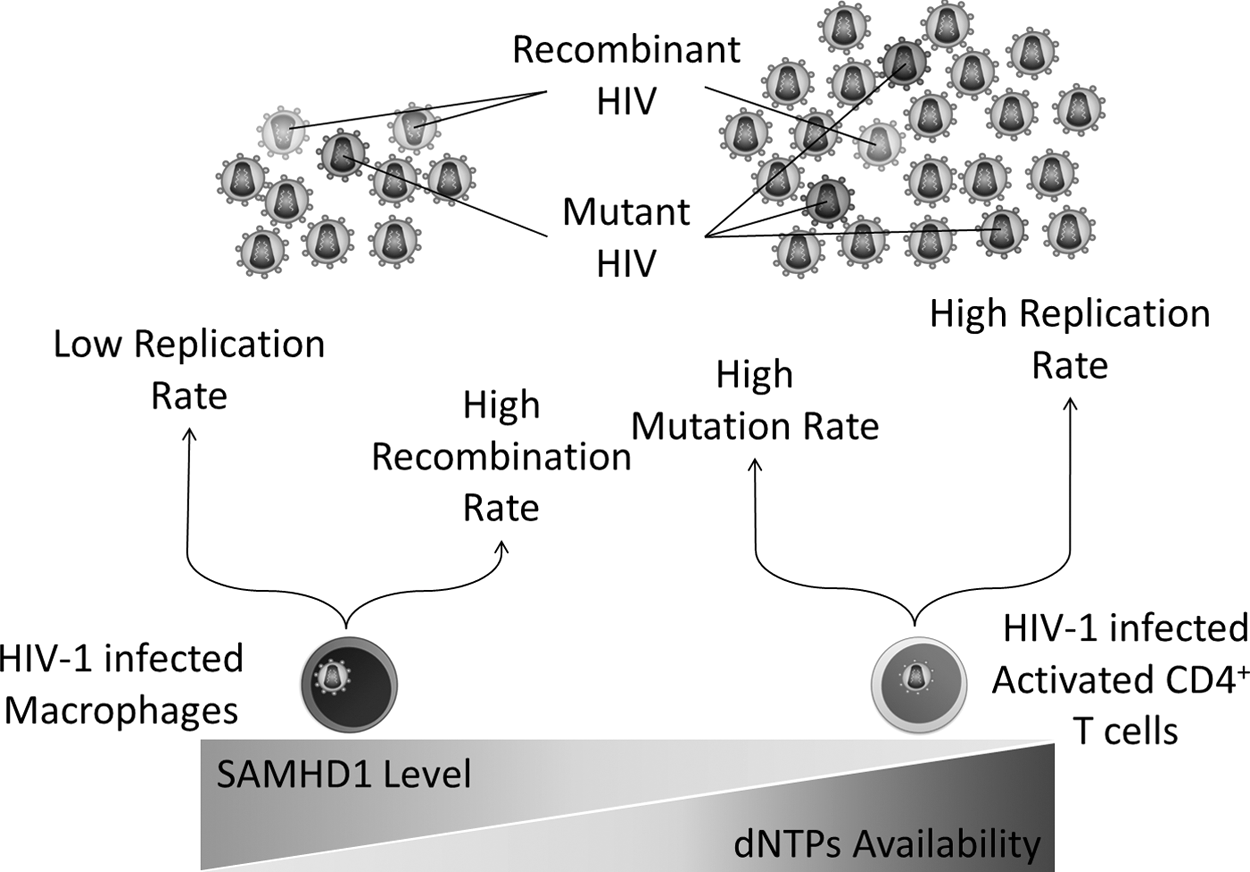
The impact of SAMHD1 and dNTPs availability on HIV-1 replication, mutation, and recombination rates. SAMHD1, sterile alpha motif and histidine-aspartic acid domain-containing protein 1.
Indeed, the availability of intracellular dNTPs is tightly regulated by the cellular mechanisms according to the cell type, activation, and proliferation status “dividing or non-dividing,” and cell-cycle phases (G1, S, G2, and M) in dividing cells. In the context of cell division, dividing cells (e.g., activated CD4+ T cells) have much higher dNTPs levels than non-dividing cells (e.g., macrophages) that range from 1–16 μM in activated CD4+ T cells and 20–40 nM in macrophages (50,53,96). This explains the higher replication and mutation rates observed in HIV-1 reverse transcripts from infected CD4+ T cells when compared with macrophages (Fig. 2) (48,53). In the context of cell-cycle phases, the G1 phase exhibits extremely very low levels of dNTPs; in contrast, the S phase remarkably exhibits very high levels of dNTPs (79), explaining why HIV-1 replication is restricted in the G1 phase (83), whereas it thrives post-S phase (112).
Ribonucleotide reductase (RNR) is the enzyme involved in the de novo synthesis of dNTPs through the reduction of ribonucleotides (50,79,119,127). Thus, the activity and expression level of RNR are known to greatly influence the intracellular level of dNTPs (127). In dividing cells, both the activity and expression level of RNR are dramatically increased before the S phase of the cell cycle to synthesize high levels of dNTPs that ensure efficient DNA replication (58,79). However, in non-dividing and resting cells (G0/G1 arrested cells), the activity of RNR has been shown to be restricted, which partially explains the low dNTPs levels in such cells (79,174).
Noteworthy, RNR activity is regulated by certain upstream cyclin-dependent kinases (CDKs), since the p21, a CDK inhibitor, can restrict the HIV-1 reverse transcription in macrophages by decreasing the level of dNTPs in a manner dependent on RNR2, a subunit of RNR, but independent on the restriction activity of sterile alpha motif and histidine-aspartic acid domain-containing protein 1 (SAMHD1) (6). Interestingly, Pauls et al. have shown that the level of p21 is strongly associated with the restriction activity of SAMHD1, even though they have not excluded the possibility of dual activity of p21 on RNR and SAMHD1 (147).
The controversy in results between the two studies could be due to the differences in the experimental system used in both studies as declared by Allouch et al. (7). Nevertheless, both studies have confirmed the efficacy of p21 to restrict HIV-1 replication in macrophages at the reverse transcription level. It is also worthy to know that upregulation of p21 in CD4+ T cells restricts the reverse transcription in HIV-1-infected activated CD4+ T cells (56), indicating that the synthesis of sufficient dNTPs by RNR is crucial for efficient HIV-1 replication. Constantly, the level of p21 has been shown to be inversely associated with HIV-1 disease progression as declared by Chen et al. (32). Hence, targeting RNR directly by nucleoside analogs and/or non-nucleoside analogs (4), or indirectly by targeting its upstream regulatory factors (i.e., p21 and certain CDKs) to reduce the dNTPs synthesis below the level required for HIV-1 reverse transcription may be considered as potential therapeutic strategies to control HIV-1 infection (56).
In contrast to RNR, SAMHD1 is responsible for dNTPs degradation into deoxyribonucleosides and free inorganic triphosphate via its dNTP phosphohydrolase (dNTPase) activity, which results in diminished intracellular dNTPs level (166). SAMHD1 is targeted for degradation in the S phase of the cell cycle to ensure an efficient DNA replication required for the cell-cycle progression (174).
Importantly, SAMHD1 has been recognized as a restriction factor for HIV-1 RT in non-dividing myeloid cells (e.g., macrophages and dendritic cells) and quiescent CD4+ T cells, but not actively dividing cells. This is mainly because dividing cells synthesize high levels of dNTPs that exceed the capacity of SAMHD1 to diminish them to levels below those required for HIV-1 reverse transcription (195). Moreover, the dNTPase activity of SAMHD1 in dividing cells has been shown to be downregulated through its phosphorylation at T592 residue via certain CDKs (179). Further, the expression level of SAMHD1 is inversely associated with the activation and proliferation state of cells (160). It is also of particular importance to know that SAMHD1 is regulated by certain CDKs, since the p21 can affect the SAMHD1 activity via targeting CDK6, an upstream regulatory of CDK2 that regulates the activity of SAMHD1 through its phosphorylation (146).
These data indicate that the restriction activity of SAMHD1 greatly depends on several factors, including: (1) the state of cell activation, (2) the levels of dNTPs (the level and activity of RNR), and (3) the phosphorylation state (phosphorylation position) of SAMHD1 mediated by certain CDKs. Moreover, the restriction activity of SAMHD1 in non-dividing cells, that is, macrophages, has been recently revealed to be highly affected by the level of cyclin L2. In other words, the level of cyclin L2 is indirectly associated with SAMHD1 level, which, in turn, indicates that high levels of cyclin L2 could support HIV-1 replication in macrophages, but not in dividing cells (104). However, further studies are required to determine the mechanisms by which the virus can affect the level of cyclin L2 in macrophages to open insights for new therapeutic targets (104). Additional possible factors may also influence the restriction activity of SAMHD1, which are still being determined (130,195).
Although SAMHD1 has been recognized as a potent restriction factor for HIV-1 in non-dividing cells, it may inversely impact immune responses and synthetic antiviral agents from different aspects. For instance, although SAMHD1 can inhibit HIV-1 reverse transcription in dendritic cells by depleting dNTPs, it may limit their capacity to activate CD4+ T cells, thus limiting the activation of potent adaptive immune responses (12). In another instance, SAMHD1 can increase the rate of HIV-1 recombination event in macrophages, thereby increasing the genetic hypervariability of HIV-1 populations that enable the virus escaping the immune responses and antiretroviral agents (see HIV-1 recombination during HIV-1 reverse transcription) (48).
Moreover, SAMHD1 partially downmodulates the efficacy of antiretroviral drugs, especially NRTIs, by decreasing their intracellular concentrations and changing their activation pathways (85). It is, therefore, not surprising to know that some investigators have proposed SAMHD1 as a possible target for inhibition (166). However, additional investigations are required to further establish whether SAMHD1 should be targeted for inhibition, activation, or both depending on the type of targeted cells.
Finally, it is worthy to point that the incorporation of dNTPs into the RT-complex in the cytoplasm of an HIV-1-infected cell is conceivable according to the scenario of reverse transcription outside the viral capsid when compared with the reverse transcription inside the viral capsid scenario (Fig. 1). Fortunately, this is no longer a barrier to explain the reverse transcription process according to the second scenario, particularly after the recent findings of Jacques et al., who revealed the ability of HIV-1 to import host dNTPs into RT-complex within the viral capsid through positively charged pores in the viral capsid, suggesting that such pores can be used as potential drug targets, particularly, because they are highly conserved across retroviruses (89).
HIV-1 Recombination During HIV-1 Reverse Transcription
Although a single HIV-1 RNA strand contains all the genetic information required for HIV-1 replication, most HIV-1 particles package two copies of the viral RNA genome (33). One of the selective benefits of this copackaging is to increase the variety of the HIV-1 genetic information via recombination (180). Genetic recombination enables HIV-1 to efficiently escape both the immune responses and antiretroviral drugs, ensuring its persistence in infected patients. In some cases, HIV-1 recombination can also rescue lethally mutated genomes, which are generated from an extremely high mutation rate (23,155).
However, for an HIV-1 recombination event to occur, at least two HIV-1 particles, each of which with a distinct homodimeric RNA genome, have to infect a single cell. These viral RNAs then must be reverse transcribed and integrated into host genome. After that, viral genes from both proviral DNAs must be expressed and one RNA strand from each of the two proviruses must be packaged into a budding virus. This new virus carrying the heterodimeric RNA genome has to infect another target cell, and HIV-1 RT has to switch the templates between these heterodimeric RNA genomes to generate recombinant reverse transcripts and then be integrated into the host genome. Finally, the recombinant proviral DNA should express its genome (Fig. 3).
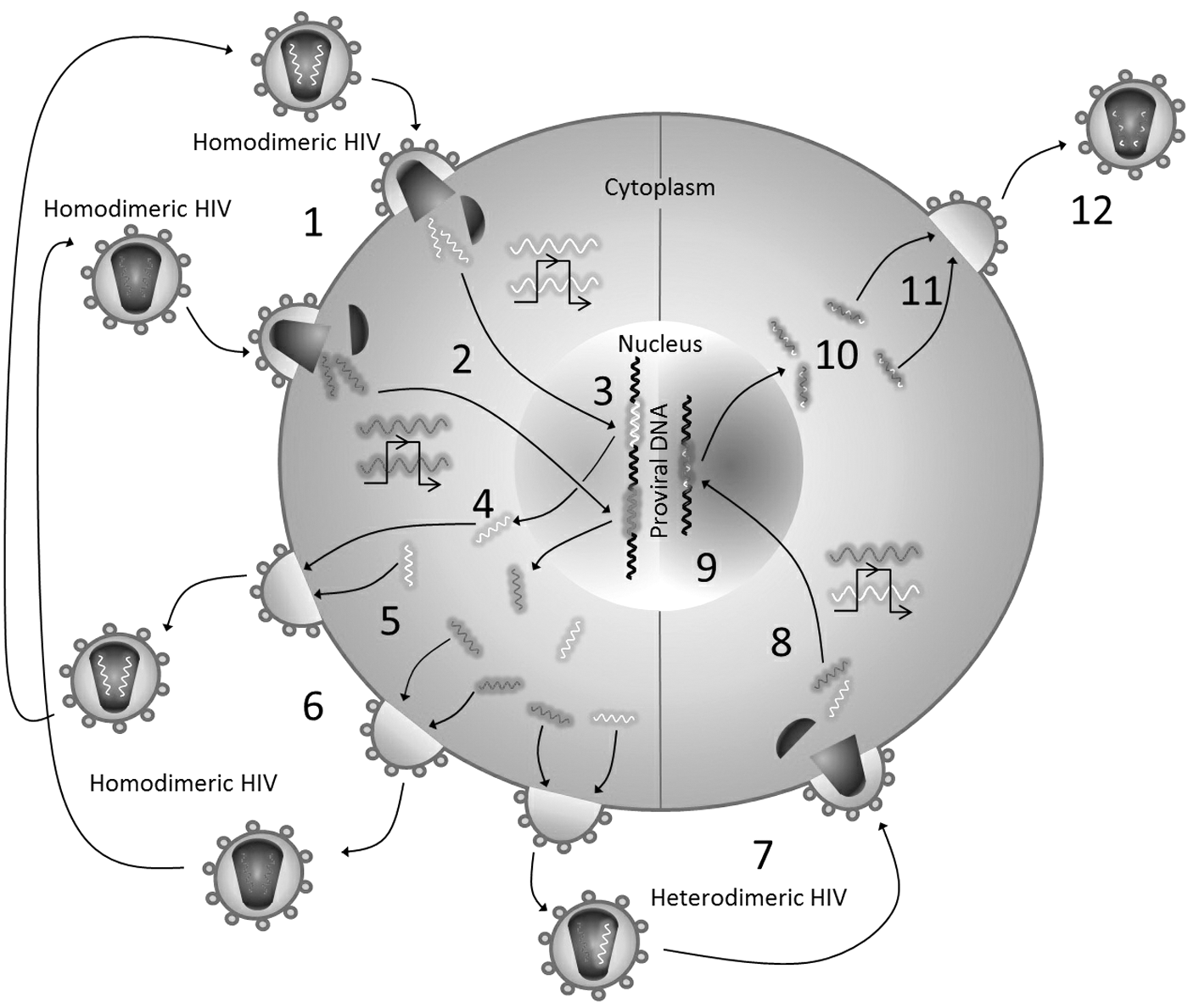
HIV-1 recombination. For an HIV-1 recombination event to occur, (1) a cell should be infected at least by two HIV-1 particles carrying distinct homodimeric RNAgenomes. (2) Each viral RNA genome must be then reverse transcribed into viral DNA. (3) Viral DNA should be integrated into host genome. (4) Proviral DNA should be expressed. (5) New viral RNA stands from each of the proviral DNAs should be transcribed and copackaged into new budding viral particles. (6) This copackaging of RNAs could result in new homodimeric HIV-1 particles carrying the same viral RNA genome, or (7) could result in new heterodimeric HIV-1 particles carrying different viral RNA genomes, or both. (8) These new HIV-1 particles carrying heterodimeric genome should infect a new target cell, and the same scenario will be repeated in steps 8–12, resulting in new recombinant HIV-1 particles as a result of the template switching of the two different viral RNA genomes during the reverse transcription process (2 and 8 steps).
It is, thus, of particular importance to know that HIV-1 recombination does not result from breakage and rejoining nucleic acid but instead results from template switching between the two copackaged viral genomic RNAs. Despite the fact that the HIV-1 recombination event requires several steps to occur, HIV-1 still has almost the highest rate of recombination events that has ever been discovered, since recombining the genetic segments of the viral RNA strands occurs ∼20% of the time throughout the HIV-1 replication (14,139). This high rate of recombination events explains the isolation of the high number of recombinant forms from HIV-1 patients, as aforementioned in the Introduction, indicating that HIV-1 recombination represents a major driver of the extensive heterogeneity of HIV-1 populations.
Copackaging of two full-length viral genomic RNAs during HIV-1 assembly/budding and template switching during the reverse transcription process are two principal events for HIV-1 genomic recombination to occur. The viral RNA packaging is mediated by the interaction of the cis-elements in the viral RNA strand (cis-acting sequences; 5′ untranslated region or 5′ UTR) with the gag-polyprotein, specifically the nucleocapsid protein (16,137,205,206). Therefore, it is not surprising to know that certain mutations in the viral nucleocapsid domain that affect its function(s) can reduce the efficiency of viral RNA packaging (74,75,205) and, consequently, affect the genomic recombination rate. Of these mutations are those that alter the two CCHC Zn2+-chelating motifs.
On the other hand, cis-elements of the viral RNA are also involved in viral genome packaging. Of these elements is the 5′ UTR, which is highly structured and forms multiple stem-loop structures (such as trans-activation region or TAR). Several studies have reported that many but not all structures of the 5′ UTR are essential for viral RNA packaging (38 –40,108,128). Another example of cis-elements that facilitates but does not directly participate in the viral RNA packaging is the Rev response elements (18,41,42,82,122,132). Besides viral RNA packaging, template switching is also central for HIV-1 recombination to occur. Thus, any factor(s) that affect(s) template switching will affect the viral genomic recombination. These data show that targeting the HIV-1 nucleocapsid protein and/or cis-elements of the viral RNA could provide a potential therapeutic approach that limits the high recombination rate of HIV-1 and inhibits its replication by generating defective HIV-1 particles that lack genomic RNA.
Importantly, the rate of HIV-1 recombination is affected by: (1) the cell type, (2) the activation and proliferation status, and (3) the level of expression of certain viral and host factors. Thus, it is logical to speculate that there would be a difference in the rate of HIV-1 recombination between dividing and non-dividing cells. In T cells, it has been reported that there are 3 to 10 recombinations/replication cycle; whereas in macrophages, it has been reported that there are around 30 recombinations/replication cycle (109,208). In line with these data, Cromer et al. results have shown that the recombination rate of HIV-1 in macrophages (6.18 × 10−3 recombination events/nucleotide/round of infection) is more than fourfold higher than in CD4+ T cells (1.46 × 10−3 recombination events/nucleotide/round of infection) (48). These in vitro genomic recombination rate estimates have been shown to be comparable with the in vivo estimates (0.5–1.5 × 10−3) of T cells but not macrophages (47).
It is also of central importance to know that most recombination events in HIV-1 patients are suggested to occur in T cells, which is, indeed, consistent with the fact that the majority of HIV-1 infection occur in T cells, as declared by Cromer et al. (47). This suggests that inhibiting HIV-1 genomic recombination in T cells could significantly lower the overall high rate of viral genomic recombination in HIV-1 patients.
However, the difference in HIV-1 recombination rates between T cells and macrophages could be attributed to the low dNTPs level in macrophages, which is associated with the delayed DNA synthesis, higher strand transfer efficiency, and increased template switching rate (48), all of which are directly associated with the HIV-1 recombination rate. These observations are also consistent with the earlier results that showed a direct association between the level of SAMHD1 and HIV-1 recombination rate (136), since the level of dNTPs is indirectly associated with the SAMHD1 level (as previously discussed) (Fig. 2). Hence, one mechanism to slow down the HIV-1 recombination rate can be achieved by targeting RNR for activation and/or targeting SAMHD1 for inhibition in HIV-1-infected macrophages; however, this is far away from rationality because it could amplify HIV-1 replication and, thus, should not be considered.
HIV-1 DNA Uracilation
HIV-1 reverse transcripts (negative viral DNA strands) are heavily uracilated (200). This event could be attributed to: (1) the low discrimination capability of HIV-1 RT between the deoxyuridine triphosphate (dUTP) and deoxythymidine triphosphate substrates during the reverse transcription process (193), which is likely to occur in CD4+ T cells and macrophages, because these cells express very high levels of dUTPs (8.4% and 12.2% of the total nucleotide pool in macrophages and peripheral CD4+ T cells, respectively) (9,183); (2) the absence of proofreading activity of HIV-1 RT; and (3) the cytosine deamination mediated by apolipoprotein B messenger RNA-editing enzyme-catalytic polypeptide-like 3 (APOBEC3) proteins, which is the major contributor to this high uracilation event during HIV-1 replication (171).
This stems from recent study results that have shown that APOBEC3 proteins play a substantial role in driving the extremely high mutation rate (∼4.1 × 10−3 per base per cell) of the virus in vivo, accounting for about 98% of these mutations compared with 2% for HIV-1 RT (50). In humans, APOBEC3 proteins belong to the APOBEC family, which contains 11 members, 7 of them belong to APOBEC3 (APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H); these proteins have a wide array of functions, including the deamination of the newly synthesized HIV-1 DNA strand (169). Of APOBEC3 proteins is the APOPEC3G, which is the first discovered host factor with intrinsic restriction activity against HIV-1 and is the major contributor to the HIV-1 DNA uracilation (17,88,168).
HIV-1 DNA uracilation could be beneficial for the virus in terms of mediation of genetic hypervariability and prevention of autointegration of the viral DNA, which limits its replication (49,200); however, the very extensive uracilation of HIV-1 DNA is harmful in terms of lethal mutations accumulation that leads to a loss of viral viability (Fig. 4). Further, prevention of the viral DNA autointegration could be considered an unwanted consequence of DNA uracilation if we considered that HIV uses this event under certain circumstances as a strategy to extend its latent reservoir in a form of pre-integration latency (170). However, the lower expression level and suboptimal activity of APOBEC3 is associated with higher viral load and rapid HIV-1 disease progression, suggesting the APOBEC3 family as a potential target for treating HIV-1 patients (49,134).
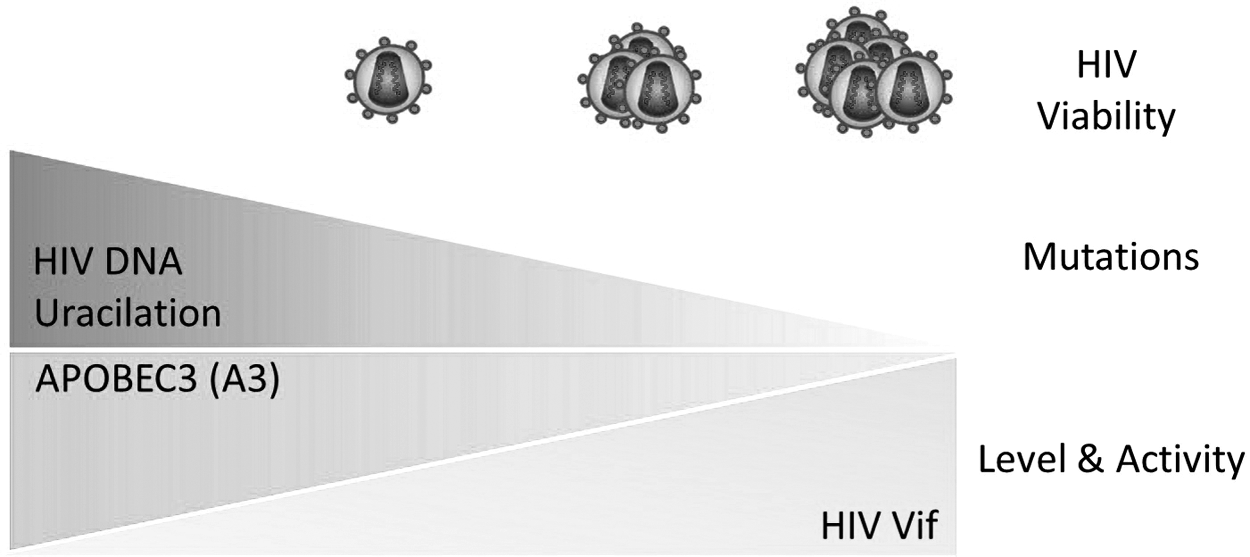
The impact of HIV DNA uracilation on viral viability. A3 proteins are cytosine deaminases that can convert cytosine bases within DNA strands into uracil bases through the deamination process. The level and functional activity of A3 proteins are directly associated with the level of HIV DNA uracilation and indirectly with the viability of HIV. HIV Vif protein can abolish the activity of A3 proteins; thus, the level and functional activity of Vif are indirectly associated with the viral DNA uracilation and, therefore, directly associated with the HIV viability.
Unfortunately, HIV-1 encodes Vif, a multifunctional protein that can efficiently abolish the function of APOBEC3 proteins by targeting them for degradation (52,134). This event seems to be dependent on the levels of Vif and APOBEC3 proteins expression. In other words, the high expression level of APOBEC3 proteins in the presence of a relatively low level of active Vif proteins drives the mediation of APOBEC3 proteins activity, and vice versa. Therefore, targeting APOBEC3 for activation and/or protection against the degradation pathway mediated by Vif-APOBEC3 axis (141) or, alternatively, targeting Vif protein for inhibition could provide potential therapeutic strategies for HIV-1 infection.
On the other hand, other studies have shown that the nuclear form of uracil DNA N-glycosylase (UNG2) could play a role in minimizing HIV-1 DNA uracilation. UNG2 is a host DNA-repairing enzyme that removes the deaminated cytosine and misincorporated uracil bases from DNA, which could render such DNA strands to be targeted for degradation by activating the host base-excision repair pathway as a result of generating abasic sites (200). Consequently, this event could affect the reverse transcription and lower the viral infectivity and replication rate. Nevertheless, HIV-1 can evade such defense mechanisms by facilitating abasic site bypass through its Vif protein (27), indicating a direct role of Vif in enhancing HIV-1 reverse transcription and replication.
The overexpression of UNG2 in a carcinoma cell line has been observed to downregulate the viral gene transcription via undetermined mechanisms, but independent on the enzymatic activity of UNG2 (61). Moreover, UNG2 counteracts the early HIV-1 replication steps in the presence of high dUTP levels (193), supporting the results that indicated that HIV-1 reverse transcripts with a high level of uracil bases tend to integrate into host genome by limiting autointegration events of the viral genome in the stage of pre-integration complex, by mediating appropriate strand transfer of the viral DNA ends, and thus promoting efficient HIV-1 replication (200). These observations suggest that UNG2 has antiviral activity to HIV-1.
Importantly, HIV-1 can escape such cellular defense responses by another multifunctional protein called Vpr. This protein can manipulate the function of host UNG2 by different mechanisms, including: targeting UNG2 for degradation through the proteasomal pathway, decreasing its expression at the gene level, or by direct inactivation (57,106,165). Vpr enhances HIV-1 replication by two- to fourfold in dividing cells (i.e., CD4+ T cells), and its expression is also essential for efficient HIV-1 replication in non-dividing cells (i.e., macrophages) (44,71). The absence of Vpr in HIV-1-infected dividing cells resulted in a fourfold higher mutation rate, whereas its absence in non-dividing cells resulted in an 18-fold higher mutation rate (34,124). This could be explained by the fact that non-dividing cells express low levels of UNG2 and high levels of dUTP (153,154). These data indicate that Vpr targets UNG2.
Although other studies have challenged this scenario of Vpr-UNG2 interaction and indicated that Vpr participates in the UNG2 incorporation into viral particles (99,153), these studies, however, are challenged by the results obtained from a previous study that indicated that UNG2 incorporation is mediated by the viral integrase (197); moreover, it is still unclear as to whether or not the virus benefits from this incorporation, since HIV-1 particles generated from cells with suppressed or lacking UNG2 (UNG2 −/−) are competent as those from wild-type cells (92).
However, these data indicate that both the viral accessory proteins, Vif and Vpr, have indirect impacts on the reverse transcription process and mediation of viral genetic hypervariability. It is also of particular importance to point out that Vif and Vpr can directly affect the reverse transcription process since they are among the proteins incorporated into the HIV-1 RT-complex. Therefore, targeting these viral accessory proteins may provide a potential strategy to combat HIV-1 infection.
Of note, if UNG2 naturally counteracts the function of the APOBEC3 family, and if the viral proteins, Vpr and Vif, can abolish the functions of these host proteins, then one could ask why HIV-1 DNA still has a high number of uracil bases. This question remains to be fully answered; however, in such a case, we would say that: First, the viral DNA uracilation is not only restricted to the APOBEC3 proteins family as already presented; as such, the capacity of UNG2 may be not sufficient to efficiently remove uracil bases from the viral DNA in HIV-1-infected cells. Second, the differences in the expression level of HIV-1 proteins, Vif and Vpr, could also affect the HIV-1 DNA uracilation process, since these viral proteins negatively impact the function and/or the level of host APOBEC3 and UNG2. Third, the type of HIV-1-infected cells (i.e., CD4+ T cells, macrophages, and dendritic cells) is another factor that affects the HIV-1 DNA uracilation, since each cell type differs in its content of dUTPs and APOBEC3 and UNG2 expression levels.
Finally, the disease stage (acute, chronic, and AIDS) could also affect HIV-1 DNA uracilation, because of the massive alteration, depletion, and dysfunction of the host cellular immune components as the disease progresses. However, additional investigations are required to study both the APOBEC3 and UNG2 in the context of HIV-1 infection at different disease stages to fully understand why HIV-1 still has high uracil bases number despite APOBEC3 and UNG2 exhibiting opposite functions in nature.
Pressure Exerted by the Immune System and Antiretroviral Therapy and the Selection of HIV-1 Escape Mutants
It has long been recognized that the immune system and antiretroviral therapy (ART) exert pressure on HIV-1, resulting in the selection of escape mutants (Fig. 5). Here, we presented a glance on the impact of such pressure exerted by certain immune system components and ART on HIV-1 hypervariability.
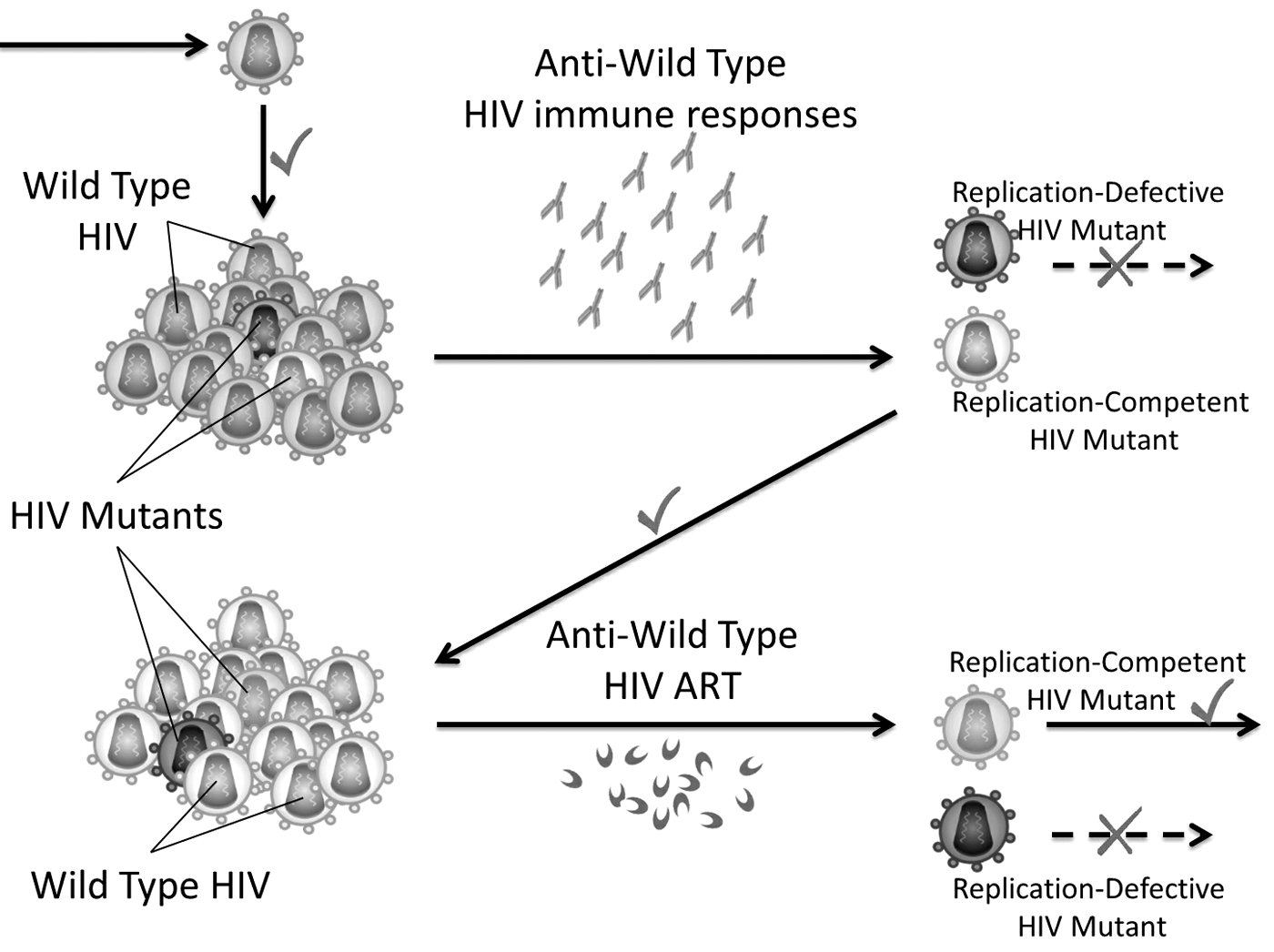
Simple schematic diagram describing HIV escape mutants' selection mediated by immune responses and synthetic antiviral agents such as ART. Right symbols (✓) = success of replication of replication-competent HIV mutant; Wrong symbols (X) = failure of replication of replication-defective HIV mutant. ART, antiretroviral therapy.
Immune-exerted pressure on HIV-1
HLA class-I-restricted cytotoxic T lymphocytes (CTL or CD8+ T cells) exerted pressure on HIV-1
A strong body of evidence supports the critical role of CTL responses in controlling HIV-1 viremia, particularly, during the early phase of HIV-1 infection, indicating that such responses could play a dispensable role in controlling HIV-1 disease progression (73). This stems from different observations that include: (1) the critical correlation between host HLA class-I alleles and set point viral load (78,152), for example, humans carrying HLA-B*57 and HLA-B*27 alleles were shown to be able to control HIV-1 infection (29,114,162,164,185), in another example, HIV-1-infected individuals carrying the HLA-B*13 allele show lower viral load and slower disease progression (84,90), and similarly are the patients carrying the HLA-B5801 allele (114); (2) the sharp increase in viral load in SIV-infected macaques with depleted CD8+ T cells (126,164); and (3) the large fitness cost observed in viral mutants that resist/escape CTL responses (65).
Indeed, CTL responses during the course of HIV-1 infection are known to play a critical role in shaping HIV-1 dynamics and evolution (28,43,76,100,131). Although strong CTL responses are exerted against HIV-1 after infection, host CTL responses fail to contain HIV-1 infection in most cases. This is because HIV-1 can quickly mitigate the potency of the host CTL responses by generating escape mutations (20,73,115). The viral adaptability to the host CTL responses is, therefore, a key determinant of HIV-1 sequence evolution in infected individuals (5,28,131). Perhaps the continuous emergence of HIV-1-resistant mutations is the most obvious demonstration for the exerted pressure by CTL responses in vivo. For instance, complete escape from HLA-B*57-restricted TW10 epitope-specific CTL can occur in about 75% of HIV-1 subtype B-infected individuals expressing HLA-B*57 that select the Gag T242 N substitution within about 3 months post–HIV-1 infection (22,107).
Despite the fact that the host-exerted pressure by CTL responses against HIV-1 results in the emergence of resistant mutants, such exerted pressure may decrease the viral replication capacity (viral fitness) and, thus, decrease the virulence of HIV-1, which, consequently, will affect the clinical course of HIV-1 infection. This is because there is a critical correlation between “HIV-1 persistence, transmission, and disease progression” and “HIV-1 fitness,” given that a small change in the viral fitness could significantly impact HIV-1 evolution, at least, because of the rapid HIV-1 replication and the nature of HIV-1 as an error-prone virus, which, in turn, could affect the pathogenesis of HIV-1 infection and transmission to new individuals (172). However, it is of considerable importance to know that fitness costs can be partially or completely restored by compensatory mutations (70,115), thereby increasing challenges for designing vaccines for HIV-1 based on T cells.
In fact, HLA class-I-restricted CTL-exerted pressure is considered a major driver of HIV-1 hypervariability at the individual- and population levels [for a review, see Ref. (28)]. These conclusive results came from statistical analyses and studying the dynamics of HIV-1 escape mutations exerted by CTL responses.
In general, the emergence of resistant HIV-1 mutants as a result of the pressure exerted by CTL can be categorized into three groups. First, antigen-processing escape mutations can obstruct the antigen processing in CD8+ T cells (e.g., B*57:03-restricted Gag A146P substitution mutation) (54); second, mutations that interfere with the binding of an epitope to HLA-I (e.g., B*27-restricted Gag R264K substitution mutation) (77); and third, mutations that abrogate or, at least, reduce the recognition of an epitope expressed on HLA class-I by some or most of host T cell receptors (TCR), also called TCR escape mutations (e.g., B*27-restricted Gag L268M substitution mutation) (87). Hence, studying HIV-1 escape mutations to understand the mechanisms and pathways involved in escape mutations emergence could help design an effective vaccine against HIV-1; therefore, additional investigations are required in this context.
Humoral adaptive immune responses (antibodies) and selection of HIV-1 escape mutants
HIV-1 env is the fastest evolving viral gene, explaining why gp120 and gp41 are the most variable viral proteins (69). HIV-1 envelope glycoproteins can exhibit about 20% of diversity in amino acids within a subtype and up to 35% between subtypes (10,101,120). HIV-1 gp120 and gp41 also represent the major targets of humoral immune responses (antibodies) (142,199). It is now well established that anti-HIV-1 antibodies exert pressure on HIV-1 and play a major role in driving HIV-1 hypervariability (198,207). Perhaps the clearest demonstration for the exerted pressure by anti-HIV-1 antibodies on HIV-1 in vivo is the continuous emergence of HIV-1 mutants that escape neutralizing anti-HIV-1 antibodies (198).
After HIV-1 infection as early as 2 weeks, specific anti-HIV-1-envelope (gp120/gp41) antibodies start appearing, which are, however, not capable of neutralizing autologous HIV-1 isolates (non-neutralizing antibodies) (181,182). Several months up to a year post–HIV-1 infection, autologous neutralizing anti-HIV-1 antibodies start appearing; however, HIV-1 results in the rapid emergence of new resistant mutants (111,133). Two to 4 years post–HIV-1 infection, extremely potent and broadly neutralizing anti-HIV-1 antibodies can be developed in a small group of HIV-1 patients (24,55,102,145,161). Although these antibodies can neutralize a wide range of heterologous HIV-1 variants, they still cannot control HIV-1 viremia from whom these antibodies were isolated, indicating that antibodies exert pressure on HIV-1 to select resistant mutants in vivo (60,102,121).
Studies have shown that infection with the homogeneous HIV-1 population (have <1% diversity in envelope variants) results in up to 10% envelope diversity during the chronic phase of infection (167), supporting the fact that antibodies exert pressure on HIV-1 and leading to the emergence of new resistant mutants. In line with these data, Wu et al. (198) have used single-genome amplification and DNA Sanger sequencing approach to characterize the circulating HIV-1 envelope variants that coexisted with a broadly neutralizing anti-HIV-1 antibody (VRC01), which targets the CD4 binding site (CD4bs) on the viral envelope gp120, to understand how the HIV-1 env gene evolves under the selection pressure of VRC01 antibody. Their results indicated that VRC01 exerts a strong pressure on CD4bs (198).
Interestingly, after a comprehensive study of several potent and broadly neutralizing anti-HIV-1 gp120 antibodies such as NIH45-46G54W, VRC01, VRC03, PG9, PG16, PGT128, etc., Bouvin-Pley et al. have shown that HIV-1 gp120 variants drift toward higher resistance to the extremely potent and broadly antibodies during the HIV/AIDS epidemic course (over a period of two decades) (21). Their results also confirmed the ongoing adaptation of HIV-1 to the humoral immune responses of HIV-1 patients. Although HIV-1 envelope developed as a result of high mutation rates can escape neutralizing antibodies, the exerted pressure by antibodies has fitness costs (122).
For this reason, Lynch et al. have evaluated the impact of such pressure exerted by broadly neutralizing anti-HIV-1 antibodies on the efficiency of HIV-1 replication (121). Their results indicated that resistant HIV-1 mutants that escape broadly neutralizing anti-HIV-1 antibodies of the VRC01 class were associated with a reduced viral replication, in part, as a result of reduced viral entry mediated by the CD4 molecule. However, this reduction in viral replication can be fully restored by compensatory mutations as demonstrated by Lynch et al. (121), which, in part, explains the limited effect of broadly neutralizing anti-HIV-1 antibodies on the viral control during the natural infection.
Helper T lymphocytes (TH cells or CD4+ T cells) and selection of HIV-1 escape mutants
Although the impact of CD4+ T cell responses in driving HIV-1 hypervariability is less established when compared with CTL and antibody responses, indeed, recent advances have revealed such an impact for CD4+ T cells. For example, studies on controller SIV-infected macaques have revealed for the first time that CD4+ T cells can exert selective pressure on the virus in vivo under low viremia (25). This notion came from studying the capacity of cytolytic Gag-specific CD4+ T cells to mediate lysing of infected macrophages during viral control (low viral load) and post-control loss (high viral load). More recent studies have indicated that HLA class-II associated HIV-1 polymorphisms can predict escape from CD4+ T cell responses, indicating that CD4+ T cells can drive HIV-1 hypervariability (59). However, additional investigations are required to further establish the impact of CD4+ T-exerted pressure on HIV-1 hypervariability.
Taken together, these data indicate that the host immune responses exert pressures on HIV-1 and contribute to the viral hypervariability by selecting resistant mutants.
ART Exerted Pressure on HIV-1 and Transmitted Drug Resistance
In light of current available HIV-1 therapeutics, around 40 antiretroviral drugs have been approved as therapeutic agents that target HIV-1 RT, integrase, protease, and viral entry and fusion, since the discovery of HIV-1 (51). This high number of antiretroviral therapeutic drugs, in part, represents a clear demonstration for the exerted pressure by ART on HIV-1 in vivo that selects resistant mutants. ART resistance has been frequently reported in HIV-1 patients, especially, when ART was used as a monotherapy. Given that, ART-resistant HIV-1 mutants' emergence is well recognized to be a major determinant factor of treatment failure in ART-treated HIV-1 patients (138,148,151).
To solve this problem, two decades ago, different drug classes of ART were combined to avoid single-drug resistance (combination strategy). Theoretically, it seemed unlikely that new resistant HIV-1 mutants will emerge against more than one class of antiretroviral drugs simultaneously (multi-drug resistance). Nevertheless, clinical investigations have demonstrated that the emergence of multi-drug resistance is common in HIV-1 patients, reflecting the high genetic hypervariability of this virus, posing a real challenge against ART success to efficiently treat HIV-1 patients (11,80,113,118,138,151,187,190,209).
Although the high genetic hypervariability of HIV-1 populations stands in front as the underlying cause of multi-drug resistance emergence, the suboptimal combined ART regimens can also contribute to ART-resistance emergence in HIV-1 patients (118). In an attempt to limit this event, practicing HIV-1 genotyping (via genotypic resistance assays) before ART initiation or regimen changes was suggested (59,118,168). Moreover, it has been recently revealed that long-term ART without virological monitoring is associated with the accumulation of mutations and emergence of ART resistance that limit drug options for the next ART regimens (10,80,113,190).
Noteworthy, transmission of ART-resistant HIV-1 mutants to other individuals restricts the ability of the same used ART regimen to control newly HIV-1-infected patients, which, subsequently, may lead to treatment failure (15,173). This phenomenon is known as transmitted drug resistance and is still a global problem that faces ART success.
Interestingly, it has been shown that newly HIV-1-infected individuals in high-income countries have higher trends (7–17%) to have at least one major drug-resistant HIV-1 mutation compared with the middle- and low-income countries (7%) (148). Nevertheless, the impact of this phenomenon can slightly affect the HIV-1 patients in high-income countries, since the viral genotyping is a standard practice before the initiation of ART in these countries. HIV-1 genotyping, in turn, helps determine optimal combined ART regimens, which, in turn, increase the rate of successful treatment among these patients (148). By contrast, the absence of viral genotyping in the middle- and low-income countries may lead to suboptimal combined ART regimens, which, in turn, increase the rate of multi-drug resistance emergence, and thus failure to control viremia by the used ART regimens (117,148).
This necessitates continuing research to develop new treatments, which is a time-, effort-, and money-consuming process. Therefore, extending the viral genotyping into middle- and low-income countries may partially solve the problem of emergence of drug resistance by improving combined ART regimens and, thus, maximizing ART efficacy (Fig. 6). In parallel, investigators are recommended to focus their efforts on developing new classes of drugs that target HIV-1 regardless of its hypervariability through targeting highly conserved viral proteins and/or host factors involved in its replication pathways (as aforementioned). If successful, this would change the way to manage HIV-1 infection.
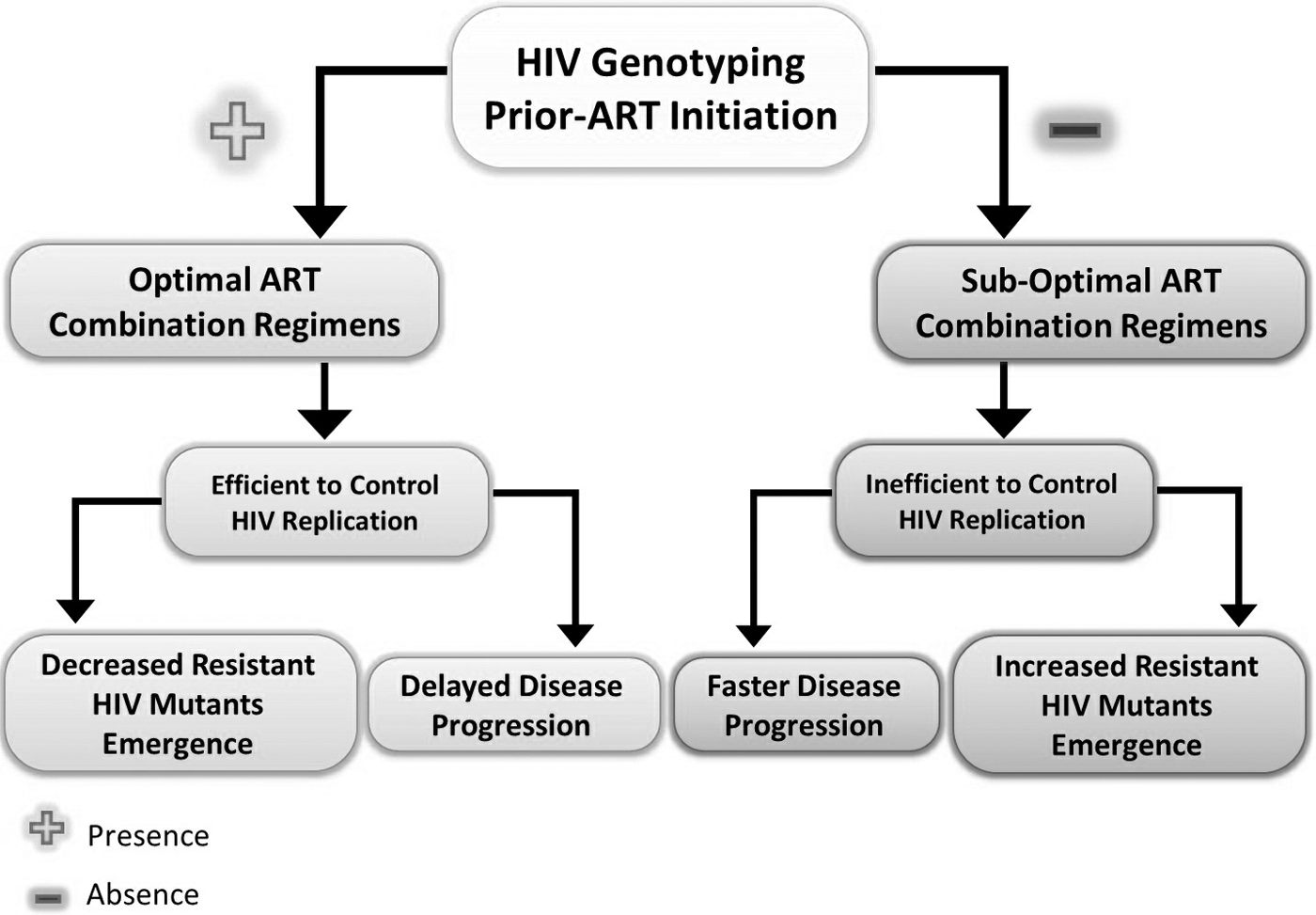
The impact of HIV genotyping before ART initiation on controlling HIV, resistance emergence, and disease progression.
Conclusions
HIV-1 extensive hypervariability is one of, if not, the most critical factor(s) that make(s) the development of preventive and therapeutic vaccines against HIV-1 infection very difficult and elusive. Moreover, it is a major barrier against the success of currently available ART. Here, we presented the most important factors that drive such extensive hypervariability in HIV-1 populations that include: first, the low fidelity nature of HIV-1 RT, which can be affected by different factors such as: (1) mutations within the viral enzyme itself, (2) divalent cations, and (3) availability of dNTPs. Second, the very high rate of HIV-1 recombination during the reverse transcription process is considered a major driver of HIV-1 hypervariability. Third, HIV-1 DNA uracilation by host APOBEC3 proteins family is another critical factor that drives extensive HIV-1 hypervariability. Fourth, the pressures exerted by the immune system and ART can also contribute to HIV-1 hypervariability by selecting escape mutants.
Although the emergence of HIV-1 mutations enable it to efficiently escape the antiviral attacks of the immune responses and ART, certain mutations can reduce HIV-1 replication capacity (fitness costs). However, the presence of compensatory mutations can mitigate the viral fitness costs.
In fact, certain HIV-1 proteins evolve faster than others; thus, targeting highly conserved viral proteins and/or host factors involved in the viral replication could be the rational way to effectively control HIV-1 infection. For example, positively charged pores in the viral capsid are supposed to be a potential drug target as has been recently suggested by a study published in Nature (89), at least, because these structures are highly conserved among primate retroviruses. Further, targeting these viral structures could limit the very high viral replication rate by limiting the incorporation of dNTPs to the reverse transcription complex inside the viral capsid. Subsequently, this can limit the very high HIV-1 mutation rate, since the mutation rate is directly associated with the replication rate.
In another example, targeting multifunctional HIV-1 accessory, Vif and Vpr, proteins can significantly affect HIV-1 replication and mutation rates because these viral proteins are directly or indirectly involved in the reverse transcription process. Moreover, Vif can abrogate the functions of host APOBEC3 proteins, which are known to express very potent anti-HIV-1 responses. Alternatively, targeting APOBEC3 proteins for activation, particularly the APOBEC3G, can significantly reduce HIV-1 viability by mediating lethal mutations. Similarly, targeting HIV-1 gag-polyproteins, particularly HIV-1 nucleocapsid protein, and/or cis-elements of the viral RNA could provide a potential therapeutic approach that limits the high recombination rate of HIV-1 and inhibits its replication by generating defective HIV-1 particles that lack genomic RNA.
Of note, a combination of different therapeutic targets could be much more efficient to combat HIV-1 hypervariability than using a single target for those seeking to develop effective therapeutic strategies.
Footnotes
Acknowledgment
This work was supported by the Deanship of Research at Jordan University of Science and Technology (Grant No. 8/2016).
Author Disclosure Statement
No competing financial interests exist.