
Abstract
Immunological memory is elicited after either vaccination or natural exposure to a pathogen and is essential for protection against re-exposure. Despite its critical importance, the ability to interrogate the veterinary animal memory immune response has long been hindered by a paucity of tools to assess immunological memory. As a result, the evaluation and analysis of protective immune responses that predict immune protection in food and fiber animals and facilitate vaccine development are obstructed. To fill this gap in knowledge in swine, we created a B cell tetramer to porcine reproductive and respiratory syndrome virus (PRRSV) nonstructural protein 7 (nsp7) to efficiently and effectively investigate the memory B cell response, a hallmark of anti-viral immunity. This novel reagent was validated by using a modified capture ELISA, tetramer pulldowns, and flow cytometry, and it was shown to detect rare, antigen-specific B cells that were present at a frequency of about 0.001% of total B lymphocytes in immune animals. The nsp7-B cell tetramer will help to characterize the PRRSV-specific memory B cell response, which is fundamentally important for understanding immunological competence and animal variation in resistance to PRRSV infection. We expect that the method will be widely applicable to the exploration of immunity to veterinary pathogens.
Introduction
T
Previous attempts to analyze the memory B cell response to vaccination or viral exposure in veterinary species have been performed by using enzyme-linked immunospot assays (ELISPOTs) (13,17). Although this is an effective test for identifying the presence of memory cells within tissues, it is limited by laborious cell isolation and culture procedures in a laboratory, and the inability to determine the frequency of memory cells in vivo due to the necessary proliferation and differentiation of memory B cells that is required to detect antibody secreting cells (ASCs) (3). In addition, the stimulation and culture of splenocytes and lymphoid tissue leukocytes include plasma cells, which may result in over-estimation of the memory cells abundance in the tissue of interest.
B cell tetramers were developed to overcome these limitations (15). Tetramers consist of four identical biotinylated proteins that are linked to a streptavidin core that is bound to a bright fluorescent protein, such as phycoerythrin (PE). Surface immunoglobulins of antigen-specific B cells bind to the proteins of the tetramer, thus enabling their detection by flow cytometry. The result is a highly sensitive reagent that is capable of capturing rare antigen-specific memory B cells using flow cytometry in whole blood or shortly after cellular dissociation from tissue (19).
Understanding the memory immune response to porcine reproductive and respiratory syndrome virus (PRRSV) is particularly important since it is the biggest threat to pig health and well-being worldwide, and it has extensive genetic plasticity. PRRSV is a rapidly mutating RNA virus that is notorious for unreliable control by vaccination. Even so, primary PRRSV infection is consistently and completely cleared from infected animals after prolonged periods by mechanisms that are not well understood (18,21). To date, the means for producing a broadly protective immune response in the pig have not been determined. The ability to efficiently and effectively analyze the porcine B cell memory response to PRRSV infection and vaccination with B cell tetramers will advance knowledge of the immunological interaction of PRRSV with pigs, and it will provide a model for similar investigations in other host-pathogen interactions.
Materials and Methods
Tissue collection
Tissues were procured via tissue sharing under the University of Minnesota IACUC protocol 1702-34568A.
Nonstructural protein 7 biotinylation and tetramer production
Soluble recombinant nsp7 protein from PRRSV2 strain VR2332 containing an amino-terminal myc tag and a carboxyl 6 × -histidine tag (referred to hereafter as nsp7) was generated in Escherichia coli bacteria and purified by cobalt-immobilized metal affinity chromatography as previously described (1,4,5,7). The molar concentration of nsp7 was determined by spectrophotometry, using an extinction coefficient of 0.036565 μM−1 cm−1 at 280 nm, with a Take3 plate and a Biotek Epoch microplate instrument.
Tetramer production was modified from Taylor et al. (19). Protein biotinylation was achieved by using an EZ-link Sulfo-NHS-LC-Biotinylation kit (Thermo Fisher Scientific) at a ratio of 1.2 biotin molecules to 1 nsp7 molecule (Fig. 1). Free biotin was removed by using 3K Amicon Ultra-0.5 mL centrifugal filters (Millipore). Biotinylation efficiency was evaluated by a quenching western blot. Ten pmol aliquots of nsp7 were incubated with twofold dilutions (0, 5, 2.5, 1.25, 0.625, 0.31, 0.156 pmol) of streptavidin (SA)-PE (Prozyme) at room temperature for 30 min. The mixtures were then run on a native protein gel for 15 min at 150 V. The gel was transferred to a nitrocellulose membrane via western blot for 1 h at 100 V. The membrane was washed with PBS and blocked for 1 h at room temperature with Odyssey blocking buffer (Licor). The membrane was washed with Phosphate Buffered Saline with Tween® 20 (PBST) and then incubated with SA-AF680 (Prozyme) at a concentration of 1:10,000 in PBST for 30 min. The membrane was washed twice with PBST and then read on a Licor Odyssey imager at an absorbance of 700 nm. The resulting level of biotinylation was determined by the reemergence of a band of biotinylated protein that was not fully quenched by the SA-PE and was able to react with SA-AF680.
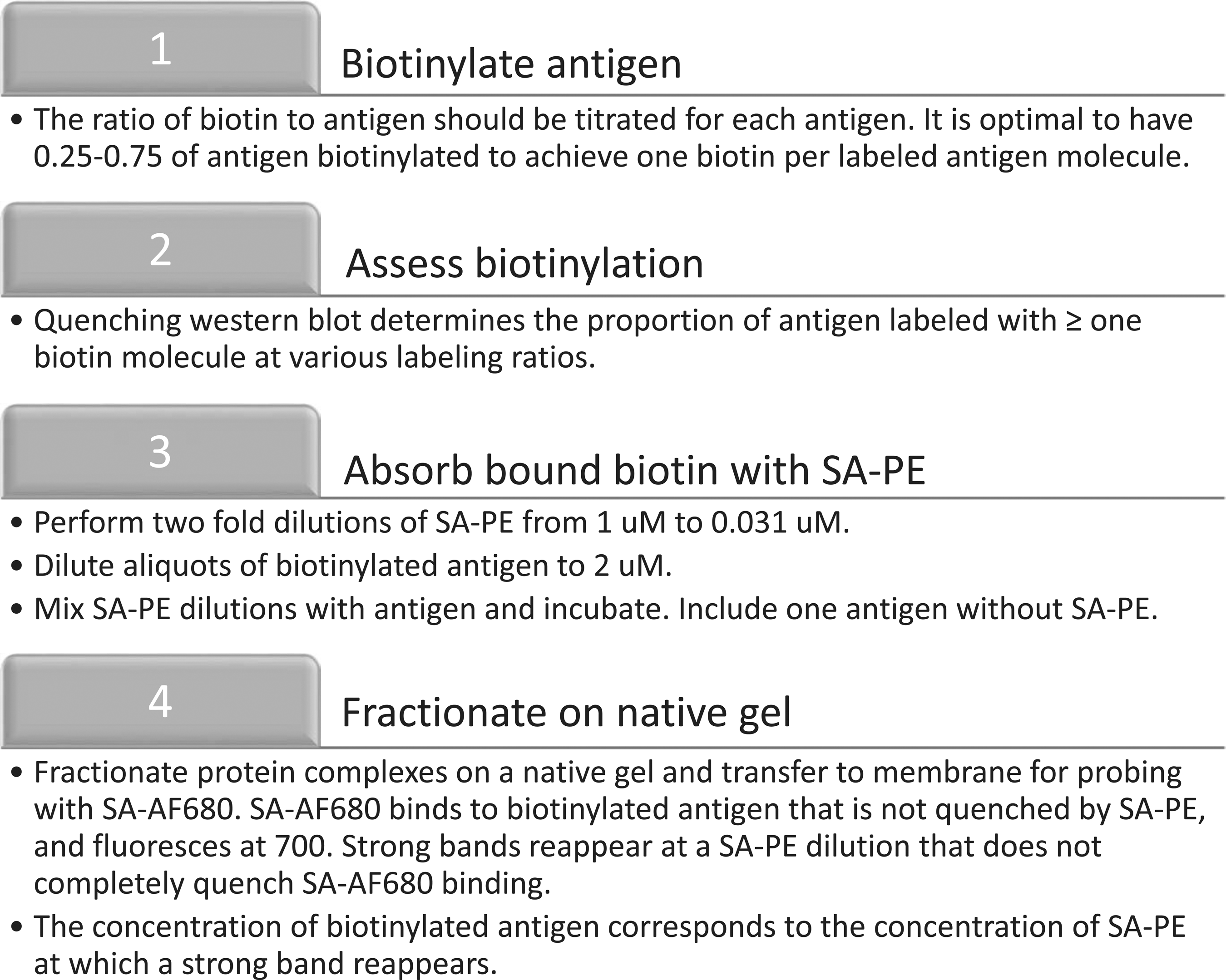
Workflow for the biotinylation of antigen. The outlined process is used to first biotinylate antigen and then to evaluate the efficiency of biotinylation.
Biotin labeling efficiency was determined based on the molarity of SA-PE when the band of biotinylated antigen reappeared, and multiplication by 4 to account for the four biotin binding sites on SA (Fig. 2). This number was divided by the molarity of the biotinylated antigen from the quenching reaction (i.e., 2 μM). Multiplying the quotient by 100 resulted in the percentage of antigen that is effectively biotinylated. A value between 25% and 75% is considered optimal. A conservative estimate of 25% was made in the example used here. In addition, the quotient was multiplied by the molarity of the biotinylated antigen, the concentration of which had been diluted to give 2 μM. The result is the molarity of biotinylated antigen in the sample and is important for tetramer formation.

Calculation of biotinylated antigen concentration. Displayed equations were used to determine the concentration of antigen that was biotinylated.
To fully arm the tetramer, four molar equivalents of biotinylated nsp7, calculated from the quenching western blot, were mixed with one molar equivalent of SA-PE, incubated for 1 h at room temperature, and finally purified with a 100K Amicon Ultra spin filter (Fig. 3). The concentration of retained tetramer was determined by measuring the absorbance at 566 nm with a Take3 plate and a Biotek Epoch spectrophotometer and adjusted by the extinction coefficient for PE. The prepared nsp7 tetramer was diluted to a concentration of 2 μM for use.

Calculation of biotinylated antigen required to fully arm tetramer. Displayed equations were used to determine the moles of antigen necessary for full tetramer formation.
A negative control decoy tetramer consisting of streptavidin-PE labeled with Alexa Fluor® 647 (AF647) (Thermo Fisher Scientific) was also created in the same manner as previously described (8,19).
nsp7 ELISA
Recombinant nsp7 was diluted in carbonate buffer, coated onto 96-well ELISA plates (Sarstedt) at 100 ng/well, and incubated overnight at 4°C. Plates were washed three times with PBS +0.05% Tween-20 (PBST) in a Biotek plate washer. Wells were blocked with 5% non-fat dry milk (NFDM) diluted in PBST, pH 9.6, for 2 h. The plates were washed and incubated with 100 μL of immune or naive serum diluted 1:50 in 5% NFDM, pH 7.4, for 1 h. Wells were washed and incubated with a 1:100,000 dilution of polyclonal goat anti-pig IgG-HRP (Bethyl) in 5% NFDM, pH 7.4, for 1 h. Plates were washed and then developed with TMB peroxidase (KPL) for 15 min. The reaction was stopped with 100 μL of 1 M phosphoric acid. Plates were read at 450 nm with a Biotek Epoch microplate instrument.
Tetramer capture ELISA
One hundred μl of serum from PRRSV immune and naive pigs was incubated with 2 pmol of nsp7 tetramer for 1 h at room temperature. Thirty μl of anti-PE microbeads (Miltenyi Biotec) was added and incubated for an additional 30 min at room temperature. Three hundred and sixty μl of PBS was added to the mixture to a final volume of 500 μl. Tetramer enrichment was then achieved by passing the mixture over an LS column in a magnetic cell separator (Miltenyi Biotec). The LS column was washed with 3 mL of PBS three times. The column was then removed from the cell separator and placed in a 5 mL fluorescence-activated cell sorting (FACS) tube. Two and one-half milliliter of PBS was added to the column, and magnetic beads bound to the tetramer were eluted from the column with a forceful plunging action. The mixture was then transferred to 2 mL Eppendorf tubes and centrifuged at 16,000xg for 15 min. The resulting brown pellet contained microbeads bound to nsp7 tetramer and bound antibodies, if present. The supernatant was discarded, and the pellet was resuspended in 1 mL of PBS. The microbead-tetramer-antibody mixture was centrifuged again at 16,000 × g for 15 min. The supernatant was aspirated, and the pellet was thoroughly resuspended in 200 μL of 5% non-fat dry milk at pH 7.4. This mixture was then analyzed for total IgG in a total IgG sandwich ELISA as previously described (17).
Tetramer pulldown
Magnetic enrichment for tetramer bound to antigen-specific cells from the tracheobronchial lymph node was carried out as previously described with modifications (Fig. 4) (8,19). Briefly, eighty million live cells were thawed from each of one PRRSV-immune and one PRRSV-naive animal. Cells were resuspended in MACS buffer (1x PBS, 2.5% FBS, 2 mM EDTA, 30 nM DNase 1) and centrifuged at 1,400 RPM for 5 min. Cells were resuspended in 100 μL of 7.5 nM decoy tetramer stain and incubated on the bench top for 10 min under foil. Cells were resuspended in 10 mL of MACS buffer and filtered through a 40 μm filter (Corning). Five hundred microliter of resuspended cells was removed for live cell count with a hemocytometer as well as flow staining for CD21 expression and viability to identify the starting number of live B cells. The remaining cells were centrifuged and then stained with 100 μL of 7.5 nM nsp7-tetramer stain. Cells were incubated for 30 min at 4°C. After incubation, cells were washed with 10 mL of MACS buffer and then resuspended in 200 μL (160 μL MACS, 40 μL microbeads) of microbead mix (Miltenyi). Samples were incubated for 30 min at 4°C. Cells were washed with 10 mL of MACS buffer, centrifuged, and resuspended in 500 μL of MACS buffer. This cellular suspension was dispensed over a 40 μm filter into an LS column (Miltenyi) resting in a QuadroMACS separator. The column was washed three times with 3 mL of MACS buffer. After washing, the column was removed from the separator and placed in a 5 mL FACS tube. Three milliliter of MACS buffer was added to the column and forcibly plunged into the FACS tube to elute cells from the column. Eluted cells were centrifuged and stained for CD21 expression and viability. Cells were analyzed on an LSRII flow cytometer (BD) and evaluated with FlowJo v10 software (Tree Star).
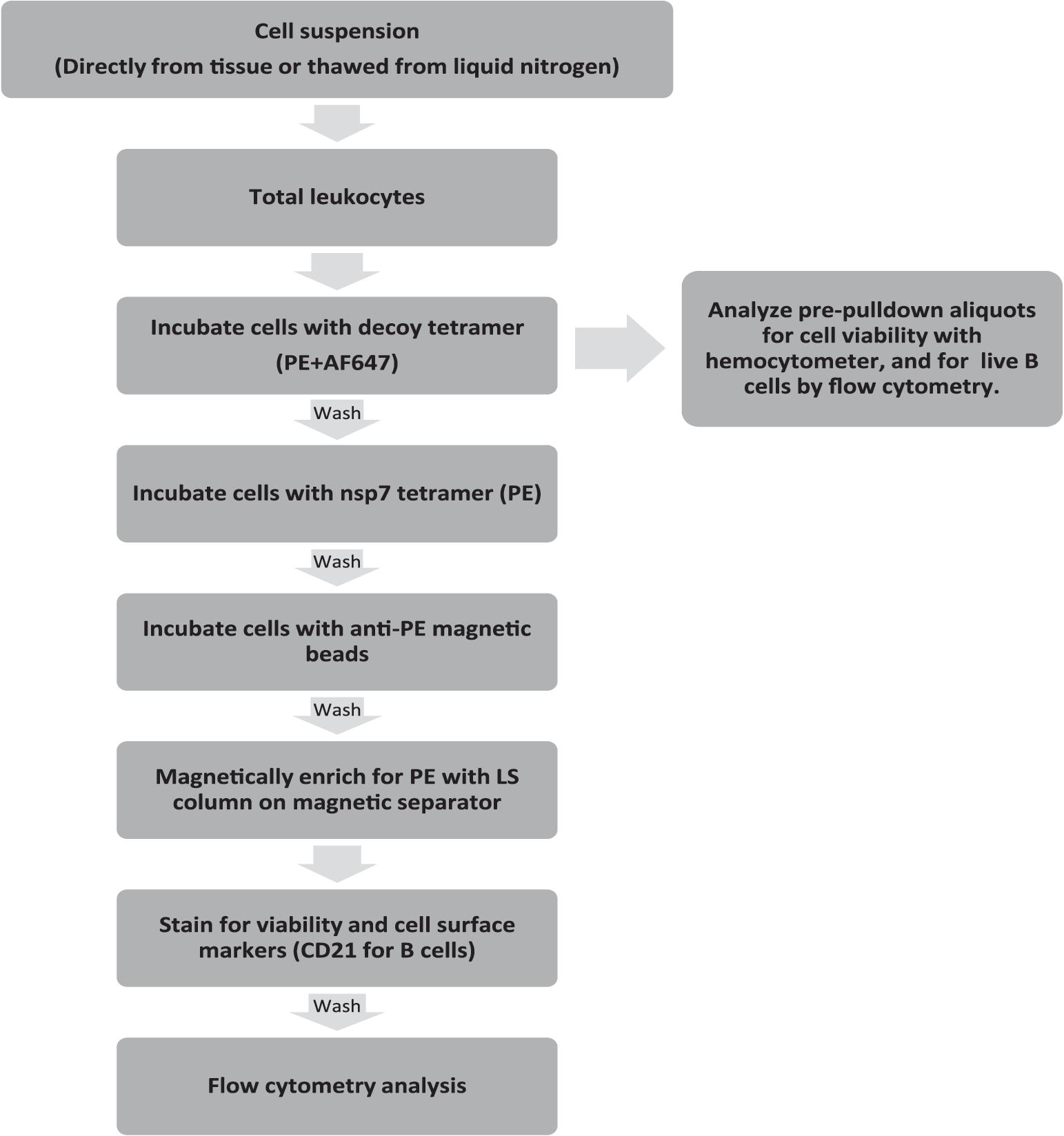
Tetramer pulldown workflow. The displayed protocol was used to identify and enumerate nsp7-specific live B cells as well as to determine the total number of starting live B cells. nsp7, nonstructural protein 7.
Results
Antigen production
Nsp7 was expressed at high levels in E. coli Rosetta cells. Three hours after isopropyl β-D-1-thiogalactopyranoside (IPTG) induction, nsp7 appeared to account for >50% of total cellular protein, and it was homogeneous on a sodium dodecyl sulfate (SDS)-polyacrylamide gel after cobalt metal affinity chromatography (Fig. 5A). Purified nsp7 was highly antigenic and specifically bound by antibodies from PRRSV-immune serum but not from PRRSV-nonimmune serum (Fig. 5B).
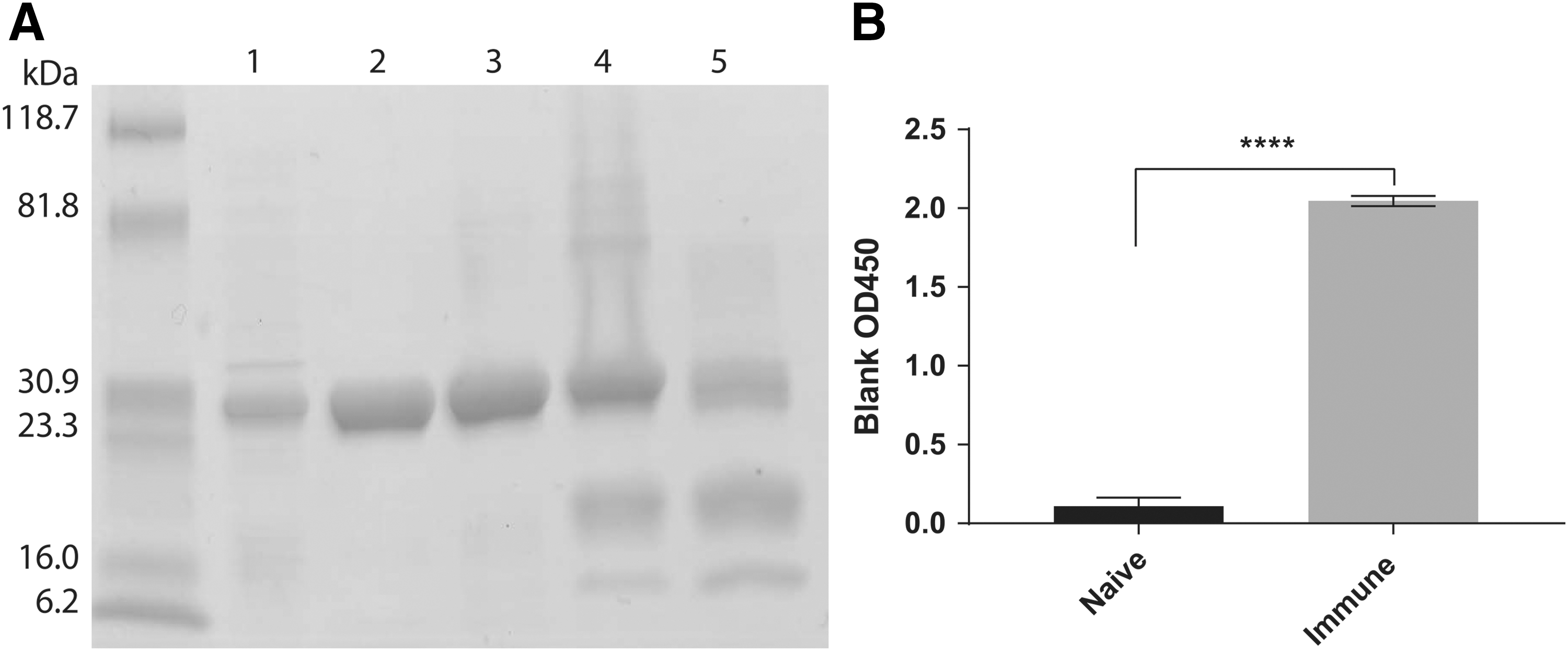
Recombinant nsp7 characterization.
Antigen biotinylation
Quantitative biotinylation is required to form the tetrameric antigen complex that is key to the method. The goal is to achieve molar equivalence so that antigens contain only one biotin and minimal disruption of native antigenicity. Hence, biotin was titrated into purified nsp7 at molar ratios of 1:1 and 1.2:1 to nsp7. The labeling results were evaluated with a streptavidin quenching western blot. Efficiency was determined by first incubating a fixed concentration, 2 μM, of biotinylated nsp7 with titrated amounts of SA-PE from a saturating excess to insufficient. The reactions were electrophoresed on a non-denaturing native gel, transferred to a nitrocellulose membrane, and finally probed with the detecting reagent, Alexa Fluor 680 conjugated SA (SA-AF680). As the concentration of SA-PE in the mixture decreased, unbound biotinylated nsp7 was permitted to bind to the detecting reagent, SA-AF680, resulting in the fluorescent exposure of bands on the western blot.
At a biotin:nsp7 molar ratio of 1:1, we observed the appearance of western blot bands at low concentrations of SA-PE (0.031–0.062 μM), indicating that less than 25% of nsp7 protein was biotinylated (Fig. 6A). At a 1.2:1 ratio, the band was clearly reduced at 0.25 μM and substantially quenched at 0.5 μM, consistent with a biotin:nsp7 labeling ratio of 0.5:1.0 (Fig. 6A). The low efficiency of labeling at a 1:1 ratio of biotin to nsp7 (about 10–25%) resulted in a majority of unlabeled antigen.
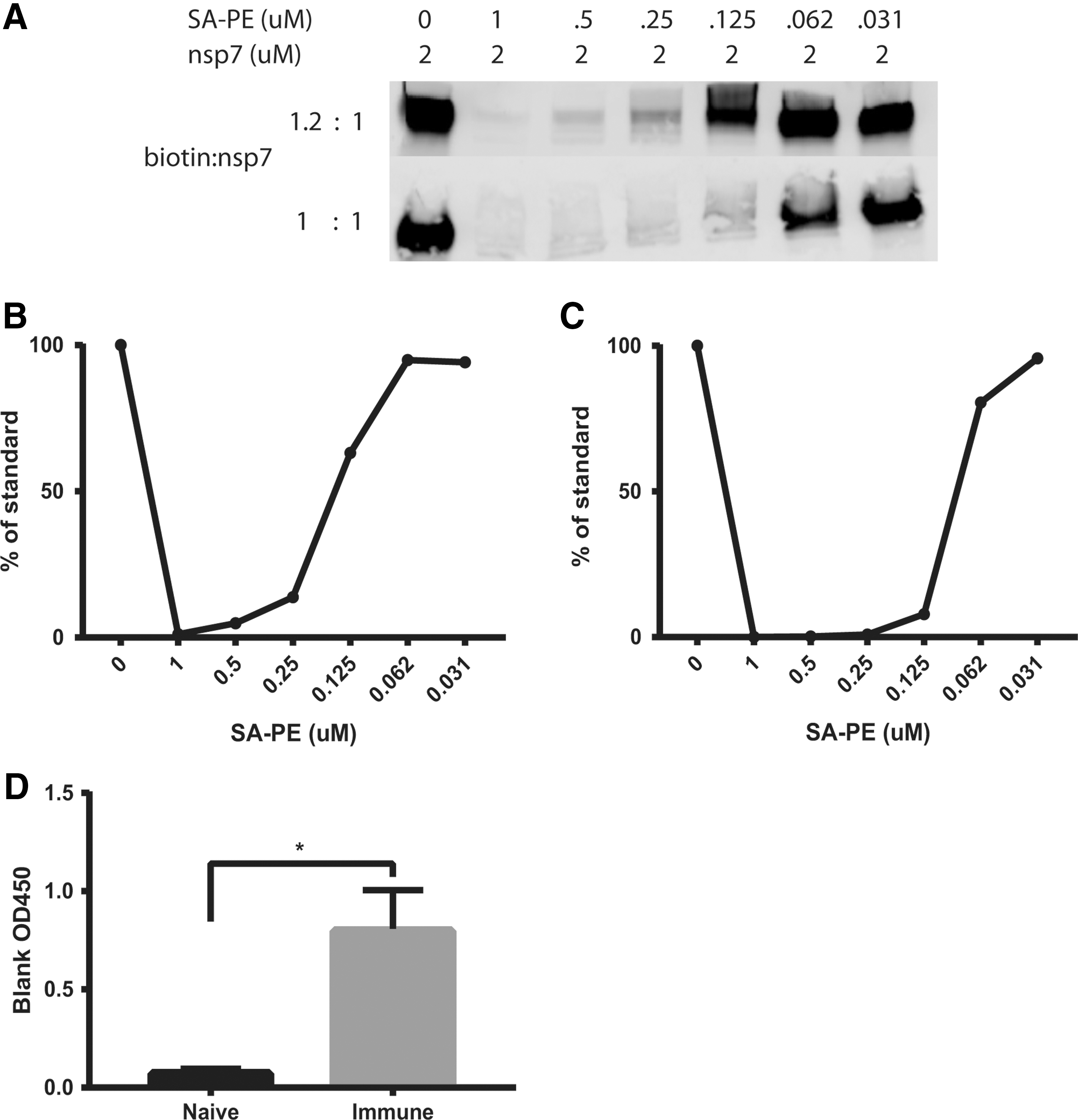
Analysis of biotinylation conditions for labeling antigen.
Antigen tetramer formation and validation
Biotinylated nsp7 was mixed with SA-PE, as described in the “Methods” section, to fully arm the tetramer. Unbiotinylated nsp7 was removed by size exclusion ultrafiltration, and the tetramer was analyzed by denaturing SDS gel electrophoresis. As shown in Figure 5, lane 4, nsp7 (30 kDa) was present. Interestingly, PE (∼250,000 kDa) and SA (∼55 kDa) do not run on SDS-polyacrylamide gel electrophoresis gels at their native molecular weights of 240 kDa and 53 kDa, respectively (Fig. 5A). Rather, PE dissociates into three subunits, α, β, and γ, with reported molecular weights of 18.3, 19.6, and 33.7 kDa, respectively (11,14). Streptavidin is a homotetramer that dissociates into four monomers that migrate at ∼12 kDa, as shown in Figure 5, lane 5 (10).
To confirm that the nsp7-tetramer retained nsp7-specific reactivity, the tetramer was incubated with PRRSV antibody positive and negative serum and anti-PE microbeads, magnetically selected on an LS column, and washed to remove unbound antibody. After elution, centrifugation, and washing of the magnetic bead pellet containing bound antibodies, the mixture was resuspended in 5% non-fat dry milk and tested in an anti-porcine IgG capture ELISA for the presence of immunoglobulin bound to the nsp7 tetramer. Evaluation of five naive and five immune animals showed that nsp7 retained specific immunoreactivity for IgG after biotinylation and tetramer formation (Fig. 6D).
Antigen-specific B cell identification
Nsp7-tetramer was evaluated for specific enumeration of antigen-specific memory B cells by using tetramer pulldown followed by FACS analysis. For tetramer pulldown, 80 million live tracheobronchial lymph node cells from a PRRSV-immune pig, and from one PRRSV-naive animal of the same age, were thawed from liquid nitrogen, stained with the decoy tetramer, and SA-PE conjugated to AF647, to detect B cells that bind common components of the tetramer, such as SA or PE, but not nsp7. Next, cells were stained with the nsp7-tetramer, followed by anti-PE microbeads, which bound PE on both the decoy and the nsp7 tetramer. After positive magnetic enrichment for PE, cells were stained for viability and CD21, a component of the B cell antigen receptor. Eluted, enriched cells from each animal were then resuspended in 425 μL of 4% paraformaldehyde and run on an LSRII flow cytometer for 8 min at identical flow rates.
Flow cytometry results were gated and evaluated, as shown in Figure 7. Analysis of the total lymphocyte populations before pulldown showed that there were more lymphocytes in the tracheobronchial lymph node of the immune animal than the naive animal (Fig. 7A). After gating for lymphocytes, single cells, and live B cells, the immune animal also had a large population of B cells bound to the nsp7 tetramer whereas the naive animal had a much smaller number of tetramer-bound cells (Fig. 7B). In addition, there were many more nsp7-specific, live B cells in the tracheobronchial lymph node of the immune animal (Fig. 7C). When controlling for the number of live B cells within the tissue, there was an ∼6-fold difference in the prevalence of nsp7-specific live B cells between immune and naive animals (Fig. 7D).
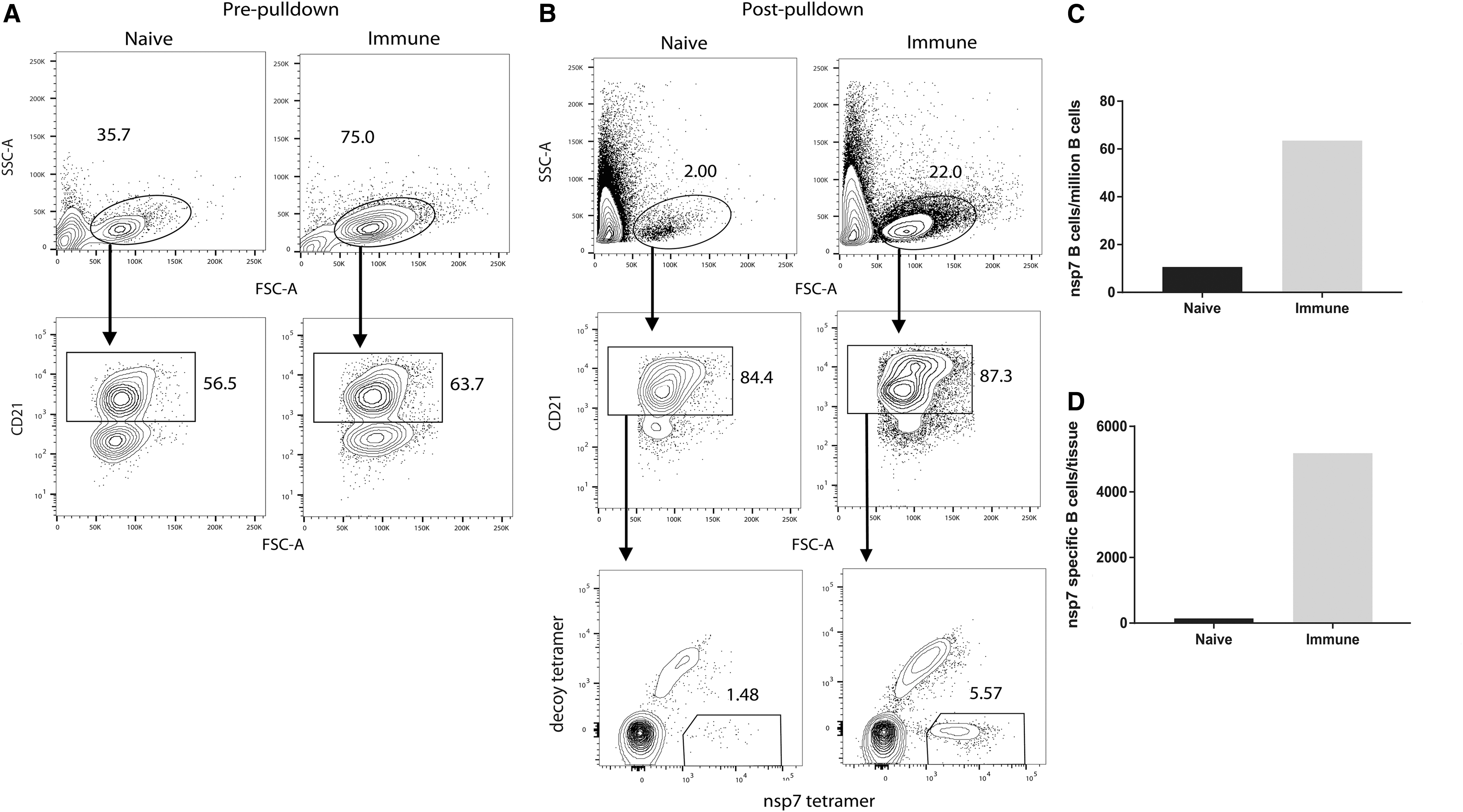
Nsp7 tetramer enumeration of antigen-specific B cells. Tracheobronchial lymph node cells from one immune and one naive animal were thawed from liquid nitrogen, counted, stained for viability and CD21 expression, and finally analyzed by flow cytometry.
Tetramer pulldown effects on live B cell numbers
Tetramer pulldowns are designed to highly enrich antigen-specific cells by selective, magnetic retention. During evaluation of its effectiveness, we noticed that there was a pronounced difference in nonspecific selected cells after pulldown, as well as in nsp7-tetramer-specific B cells, between the naive and immune populations (Fig. 7B). We also noted here, and in additional experiments, that total lymphocytes and B cell frequencies were substantially higher in lymphoid tissues from immune animals (Fig. 7A).
To determine whether immune status affected the quality of tetramer pulldown enrichment, frozen splenocytes from 30 naive and immune animals were thawed and analyzed with nsp7-tetramer pulldown. Comparison of starting and eluted live B cell numbers showed a highly significant correlation regardless of initial starting cell number, indicating that the method maintains efficiency across a wide range of starting cell population sizes (Fig. 8A). Generally, a 1,000-fold enrichment was achieved by the tetramer pulldown without a significant difference in eluted live B cell numbers between naive and immune samples (Fig. 8B).
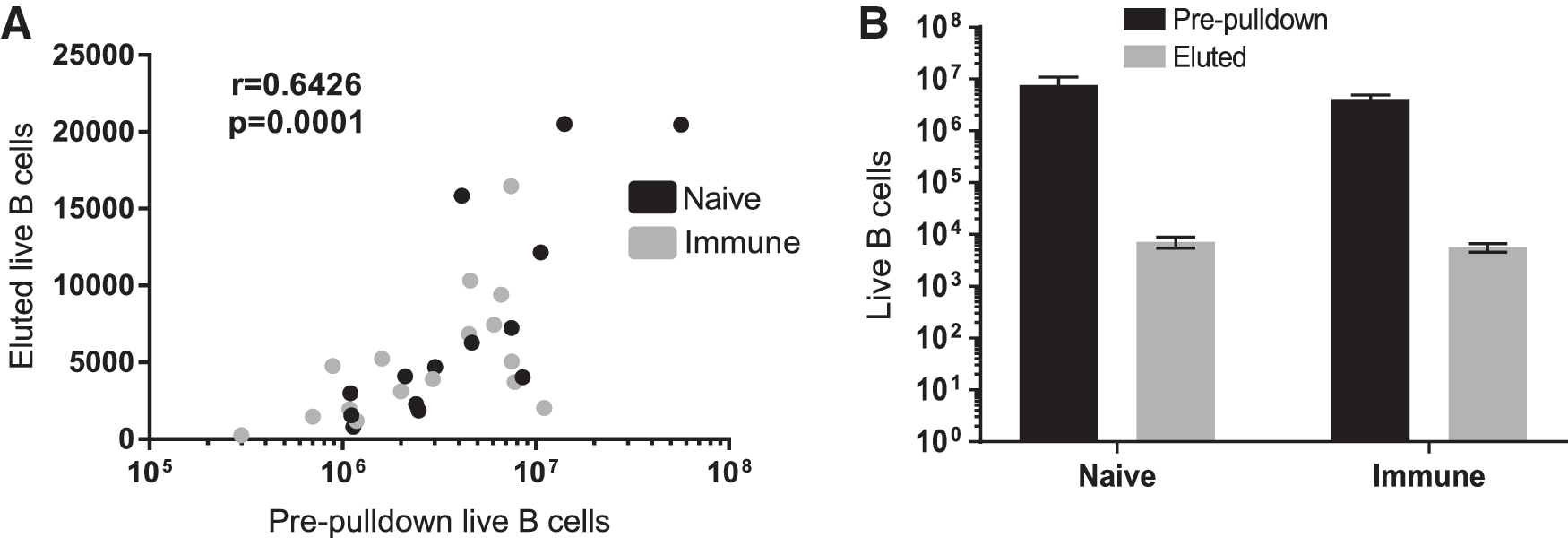
Comparison of live CD21+ B cells in splenocytes before and after tetramer pulldown.
Discussion
Limitations and restrictions on the prophylactic and therapeutic use of antibiotics for disease control, as well as emerging and re-emerging viral diseases, in food animal species provide a compelling need for immunological means of disease prevention and intervention. Vaccination is a highly effective immune countermeasure, but effective vaccines have proved difficult to develop against a number of important veterinary animal diseases. In swine alone, a wide range of bacterial pathogens, including, but not limited to Hemophilus parasuis, various Mycoplasma spp., Mycobacterium spp., Pasteurella spp., Brachyspira spp., and Bordetella brochiseptica, are not effectively controlled with existing products. With respect to viral pathogens, PRRSV vaccines are incompletely effective, whereas porcine circovirus 2 vaccines are highly effective, but the mechanism of protection is not known and new variants that are emerging raise concerns about future protection.
Memory B and T lymphocytes are the key surveillance and effector cells of adaptive immunity that are responsible for vaccine efficacy. Reagents and tools that facilitate identification, monitoring, and characterization of pathogen-specific memory lymphocytes will further a mechanistic understanding of vaccine efficacy and promote development of better products. Cell surface markers for memory B and T cells of veterinary species are sparse and, perhaps, incompletely characterized, and do not identify antigen- or pathogen-specific responses. However, tetramers are highly specific, sensitive, and biased toward detection of high-affinity IgG expressed on the surface of memory B cells. In a reversal of typical approaches to development of immune cell markers, B cell tetramers provide a route to identification and isolation of memory B cells that are then analyzed for markers of memory.
Here, development of tetramers for pathogen-specific memory B cells is described, using PRRSV in swine as a relevant example. The porcine memory immune response to PRRSV is not well characterized, resulting in a major gap in knowledge about immunological resistance to genetically diverse viruses. Previous investigations into the porcine anti-PRRSV memory immune response have utilized ELISPOT, an effective assay for enumerating ASCs (13,17). However, ELISPOT requires laborious cell culture, various conditions to differentiate plasma cells from memory cells, and is imprecise due to cell proliferation and differentiation that occurs during culture (17). Tetramers prevent these problems by their ability to bind high-affinity memory B cells that are directly isolated from animals or after recovery from frozen storage. In addition, they enable substantial characterization of antigen-specific B cell-mediated immune protection.
The creation of an effective tetramer to investigate the role of memory B cells in veterinary immunology and vaccinology starts with the production of a conserved, pure, highly immunogenic protein that is soluble in physiologically compatible solutions. In the case of PRRSV, nsp7 is known to be highly conserved among PRRSV-2 variants (2). Fortuitously, it is readily expressed at high levels in soluble form in E. coli and retains solubility after purification.
The appropriate quantitative biotinylation of nsp7 is arguably the most important aspect of tetramer manufacturing. Although the reaction itself is straightforward, the desired ratio of biotin to nsp7, that is, one biotin per molecule of protein, requires careful titration for each labeling reaction. Heavily biotinylated proteins have reduced immunoreactivity, whereas they have low levels of waste valuable reagent. Under the conditions of our laboratory, a biotin:nsp7 ratio of 1.2:1 yielded an nsp7 product in the desired ratio range of 0.25–0.75 biotin per protein molecule. After confirmation of the biotinylated nsp7 concentration and the removal of unbound biotin, the tetramer was fully armed by incubating biotinylated protein with the streptavidin-PE at a 4:1 molar ratio for 30 min at room temperature, after which remaining free nsp7 and unbound biotinylated nsp7 was removed by size exclusion centrifugation.
B cell tetramer recognition of cognate antibodies by ELISA established that nsp7 antigenic function and specificity was retained in the tetramer. The tetramer was incubated with PRRSV-naive or -immune serum, magnetically enriched to remove unbound antibody, and finally evaluated in an anti-porcine IgG capture ELISA to detect antibodies bound to nsp7 on the tetramer. To our knowledge, this is the first description of this novel assay that showed that antibodies from PRRSV-immune animals are able to specifically bind the tetramer, whereas antibodies from naive animals showed only background reactivity. Tetramer incubation with lymph node cells from naive and immune animals showed that it specifically recognized antigen-specific B cells.
The rarity of antigen-specific memory B cells in blood and lymphoid tissues, combined with the structural complexity of the tetramer, results in high nonspecific backgrounds with minimal ability to directly identify target cells. As a result of this, a decoy tetramer (PE+AF647) was first incubated with cells to identify naive B cells that are capable of binding to PE or the streptavidin core. This was followed by incubation with the nsp7 tetramer (PE). Thus, specificity was enhanced by identifying naive B cells that are capable of recognizing components of the tetramer rather than nsp7. Magnetic enrichment, referred to as tetramer pulldown in the literature, enhanced sensitivity by concentrating the number of tetramer-specific cells and allowing for the enumeration of nsp7-specific cells within millions of cells. Tetramer pulldown followed by FACS analysis enabled the detection of nsp7-specific B cells at a frequency of about 1/100,000 total B cells. Further validation of the antigen specificity of B cells isolated by antigen tetramers can be accomplished by ELISA testing of supernatants from B cells that are activated and expanded in vitro, or by ELISPOT, as described (13,17).
The tetramer pulldown enrichment step may lead to confusion in FACS data presentation and interpretation. During routine FACS analysis, cells are gated sequentially for lymphocytes, single cells, live B cells, and finally for nsp7-specific cells. The final flow plot, as shown in Figure 4, displays the decoy and nsp7 tetramer antigen specificity of all live B cells in the FACS sample. Hence, the nsp7-specific gated population shows the percentage of nsp7-specific live B cells in the sample after tetramer pulldown. The remaining, nongated cells are indicative of the amount of background non-specificity that is inherent to the pulldown method. For meaningful interpretations of antigen-specific B cell abundance and the strength of immune responses, we express the tetramer-specific cell populations as a frequency of total B cells that enter the pulldown method, or of total B cells that are present in the starting lymphoid tissue, or as an abundance in the entire lymphoid tissue itself.
The strong correlation between the starting number of live B cells in tetramer pulldown and the eluted number, regardless of immune status, shows that the method did not introduce a selection bias due to sample size or immune status. For example, beginning a tetramer pulldown with ∼6 million live B cells results in roughly 6,000 live B cells in the eluted fraction. Therefore, the number of nsp7-specific B cells that is displayed in a final flow plot is representative of the starting condition. In practice, we seek to harvest and process entire lymphoid tissues, or to determine the proportion of total tissue that is processed. The total number of live leukocytes in a tissue sample is recorded before freezing in liquid nitrogen. After thawing, we record the proportion of total tissue cells used for an experiment, assess viability, and the proportion of B cells by staining with CD21. At the end of an experiment, data can be normalized to the starting number of live B cells, or per million live B cells, or to total number in the tissue. The process removes the inherent variability that enrichment introduces and results in a more easily interpreted result.
B cell tetramers have been used in mice and humans to characterize recognition of self and foreign antigens as well as the development of the humoral immune response and memory response to innocuous antigens (6,9,12,20). This information has been profoundly important in the understanding of basic B cell ontogeny and memory development. In the pig and other veterinary species, the memory immune response to viral infection is currently a black box due to the lack of effective tools and reagents. Creation of the nsp7 tetramer will facilitate an in-depth investigation into the characteristics and quality of the memory B cell response generated to PRRSV infection and vaccination. The methods are applicable to all animal species for which relevant antigens are described or can be identified and produced in soluble form.
Footnotes
Acknowledgments
The authors thank Justin Taylor and Marc Jenkins for introduction to the methodology and advice in implementation, and Paul Champoux at the University of Minnesota flow core facility for expert technical assistance. The research was supported by USDA NIFA Grant No. 2015-06966, an NIH fellowship to MCR from the training Grant No. T32 OD010993, and an AASV Foundation grant for KG.
Author Disclosure Statement
No competing financial interests exist.