
Abstract
Understanding dengue virus (DENV)-induced hemorrhage remains a challenging jigsaw puzzle with many pieces missing to understand the complex interactions between DENV and blood coagulation system. To use flow cytometry studying the interactions between DENV and human platelet aggregation receptor, glycoprotein IIb/IIIa (gpIIb/IIIa), directly conjugated fluorochrome monoclonal antibody (mAb) is essential to facilitate multifluorochrome immunostaining. However, the obstacle was that no directly conjugated fluorochrome-anti-DENV mAb had been commercially available. To overcome, we directly conjugated fluorochrome to a primary anti-DENV mAb using a LYNX rapid conjugation kit. Flow cytometry analysis showed that this conjugated antibody and anti-gpIIb/IIIa mAb were able to detect DENV and CD41a simultaneously. Fluorescence microscopy analysis further demonstrated CD41a superficially and DENV intracellularly. Potentially, this strategy can facilitate virologists for directly conjugating any virus-specific primary antibodies, which are not commercially available with fluorochrome, to study the infectivity in any surface marker-specific hosts through flow cytometry. Together, DENV can interact with both human gpIIb/IIIa− and gpIIb/IIIa+ cells revealed by flow cytometry and fluorescence microscopy for the first time.
Introduction
D
Even though it is clear that both viral and host factors play important roles in the course of infection, a fundamental knowledge gap still remains to be filled regarding host cell tropism, crucial host immune response mechanisms, and viral markers for virulence. The pathogenesis of dengue virus (DENV) remains a challenging jigsaw puzzle with many pieces missing to understand the complex interplay of viral and host factors. Severe dengue is accompanied by hemorrhage, plasma leakage, and organ impairment. Despite intensive research, their underlying mechanisms causing severe dengue are still not well understood partly due to the lack of appropriate animal disease models and in vitro human cell models of infection (6).
Laboratory detection of DENV infection in clinical specimens has mainly involved several techniques such as infectious virus isolation, virus-specific antibody determination, and viral RNA direct detection (15). Isolating and typing the virus from patient blood have remained the “gold standard” and provides advantages over other detection methods of virus infections. However, this technique is difficult because the virus does not grow to high titers in patient blood and assays for titrating virus are time-consuming (13).
Recently, immunostaining analyzed by flow cytometry has been used to study virus-cell interaction, and it can also serve in the rapid detection of the virus-infected cells (10). Flow cytometry has emerged as a method of choice for automated analysis of cells in suspension. Flow cytometry analysis is accomplished by moving thousands of cells (per second) through a laser beam and capturing the light that emerges after each pass. This instrument permits rapid analysis at the level of single particle by optical means based upon laser activation of fluorescent dyes. Fast multiparameter examination of individual cells has made flow cytometry an invaluable tool for both qualitative and quantitative analyses. With the development of permeabilization techniques in flow cytometry and the availability of various monoclonal antibodies (mAbs) that specifically bind with cell surface and intracellular antigens, it is now possible to use flow cytometry to identify the virus-infected cells (5). The main advantage of revealing fluorescence by flow cytometry consists in the possibility to perform a large number of measures in the test sample, in an objective, rapid, and reproducible manner. Moreover, the multiparameter flow cytometry analysis of double- and triple-stained cells gives investigators the opportunity to identify which cells are infected, as well as to quantitate the number of virus-infected cells (1).
However, one important obstacle of intracellular DENV detection by flow cytometry is that there is no commercial directly conjugated fluorochrome anti-DENV mAb. Most researchers necessarily use commercially available mouse mAb to detect DENV infection by indirect immunostaining method. However, its disadvantages include the potential for cross-reactivity and the need to find primary antibodies that are raised in different species or of different isotypes when performing multiple-labeling experiments. Mouse primary mAb can be used for detecting only one marker in each detection if secondary conjugated fluorochrome anti-mouse immunoglobulin mAb is applied. Thus, directly conjugated fluorochrome antibody is essentially needed for simultaneously detecting more than one marker in the same cell.
The objective of this study was to develop a simple method for directly conjugating an interested fluorochrome to commercially available mouse primary anti-DENV mAb. The conjugated antibody established in this study was first tested to detect intracellular DENV inside the infected well-known host of DENV Vero cells. Afterward, the antibody was further tested for simultaneous detection of intracellular DENV and surface marker of megakaryocyte by using MEG-01 cells as a model.
Materials and Methods
Immunostaining
MEG-01 cells or Vero cells were adjusted to 1 × 105 cells and washed with PBS once. The surface glycoprotein IIb/IIIa was stained by incubating the cell with anti-human CD41a-FITC (fluorescein isothiocyanate) (BD Pharmingen) at 4°C for 30 min in the dark either before or after cells being fixed and permeabilized. Intracellular DENV was always stained after cells being fixed and permeabilized. The cells were washed once with 1 mL of PBS and fixed with 1 mL of 4% paraformaldehyde in PBS at 25°C for 20 min. The fixed cells were washed once with 1 mL of PBS and permeabilized with 1 mL of BD Perm/Wash® buffer (BD Pharmingen) at 25°C for 20 min. The cells were blocked by adding either PBS or 3% bovine serum albumin (BSA) (Sigma-Aldrich). Finally, 1 μL of PE-anti-DENV antibody was added into the cell pellets followed by incubating at 25°C for 1 h. After incubation with the antibodies, the cell pellets were washed twice with 1 mL of PBS and fixed with 1 mL of 1% paraformaldehyde in PBS. The staining was applied to uninfected cells in parallel with DENV-infected cells.
Flow cytometry analysis
The stained cells were analyzed by FACSCalibur (Becton Dickinson) using CellQuest Software (Becton Dickinson). Dead cells and cell debris were excluded from analysis using FSC versus SSC double dot. No-stained cells were used to determine the compensation and cutoff cells without CD41a. Uninfected cells were used to determine the compensation and cutoff cells without DENV.
Fluorescence microscopy analysis
After double fluorescence staining, the cells were centrifuged at 2,000 rpm for 5 min. The supernatant was discarded. The cells were mixed with the remaining supernatant at the bottom of tube. The cells (10 μL) were dropped onto a microscopic slide with its cover slip. The slide was visualized under a fluorescence microscope using the B-2E/C filter for FITC and G-2A filter for PE. The cell pictures were captured and merged using NIS Element D4.10.00 software.
Cell culture
The human megakaryoblastic cell line MEG-01 was purchased from the American Type Culture Collection (ATCC) and cultivated in RPMI 1640 (GIBCO) supplemented with 10% FBS (GIBCO), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin as the maintenance medium, at 37°C in a 5% CO2 humidified atmosphere. Cell numbers were maintained below approximate density of 106 cells/mL by refreshing a new medium twice or thrice per week.
DENV infection
MEG-01 cells were adjusted to 3 × 105 cells and washed with PBS. The cells were incubated with various amounts of DENV, including 0.5, 1, and 2 mL of 3 × 105 focus-forming unit (FFU)/mL for a multiplicity of infection (MOI) of 0.5, 1, and 2, respectively. MEM containing 2% FBS was used as negative control (uninfected cells). All the cultures were mixed and incubated at 37°C, 5% CO2, for 2 h. The infected cells were washed twice with PBS and fresh RPMI-1640 was added for further culture.
DENV production
Vero cells were maintained at a concentration of 1 × 106 cells/plate in Minimum Essential Medium (MEM) (GIBCO) containing 10% FBS at 37°C, 5% CO2. DENV type 2 (strain 16681) was added to 80–90% confluent cells at an MOI of 0.1 and incubated at 25°C with rocking for 2 h. After incubation, the culture medium was replaced with fresh MEM containing 2% FBS and cultured at 37°C, 5% CO2. The medium of DENV-infected Vero cells was collected and replaced every 3 days. The collected medium was centrifuged at 1,500 rpm, 4°C, for 5 min. The supernatant was aliquoted and stored at −80°C until use.
DENV focus-forming assays
For quantification of the concentration of DENV, Vero cells were grown to be a confluent monolayer in MEM containing 10% FBS in T75. The DENV was serially 10-fold diluted until 106 in MEM containing 2% FBS and then inoculated into the cultured cells and incubated at 37°C for 1 h. After that, the DENV-cultured medium was replaced with MEM containing 2% FBS and 2% methylcellulose (Sigma-Aldrich) and kept at 37°C for 5 days. DENV foci were detected by immunohistochemistry and the viral foci were counted microscopically.
Results
Direct conjugation of fluorochrome to DENV primary antibody
To directly conjugate fluorochrome to anti-DENV primary antibody, we used the LYNX Conjugation Kit from AbD Serotec (LNK021RPE) for conjugating phycoerythrin (PE) to the mAb, clone D3-2H2-9-21 from Millipore (MAB8705). This antibody specifically reacted to all four DENV serotypes. First, we added 1 μL of the modifier reagent for every 10 μL of the primary antibody sample and mixed gently. Second, we pipetted the mixture onto the LYNX lyophilized, mixed gently, and incubated in the dark at room temperature (20–25°C) for at least 3 h. Third, we added 1 μL of the quencher reagent for every 10 μL of the primary antibody used and incubated for 30 min before use. The conjugated antibody was kept at 4°C until use. In conclusion, pipettes were used thrice and the overall incubation time was 3 h 30 min.
Single detection using directly conjugated PE-anti-DENV antibody
To test the capability of directly conjugated PE-anti-DENV antibody for staining intracellular DENV, this antibody was first tested to detect DENV in DENV-infected Vero cells. This cell is one of the most common cell lines for DENV isolation and propagation. Vero cells were infected with DENV at an MOI of 0 (uninfected) and 0.1. At 3 days postinfection (dpi), the cells were stained with 2 μL of the conjugated antibody. Flow cytometry analysis showed three obviously distinguished fluorescence-intensity peaks of no-stained, uninfected and DENV-infected Vero cells (Fig. 1B).
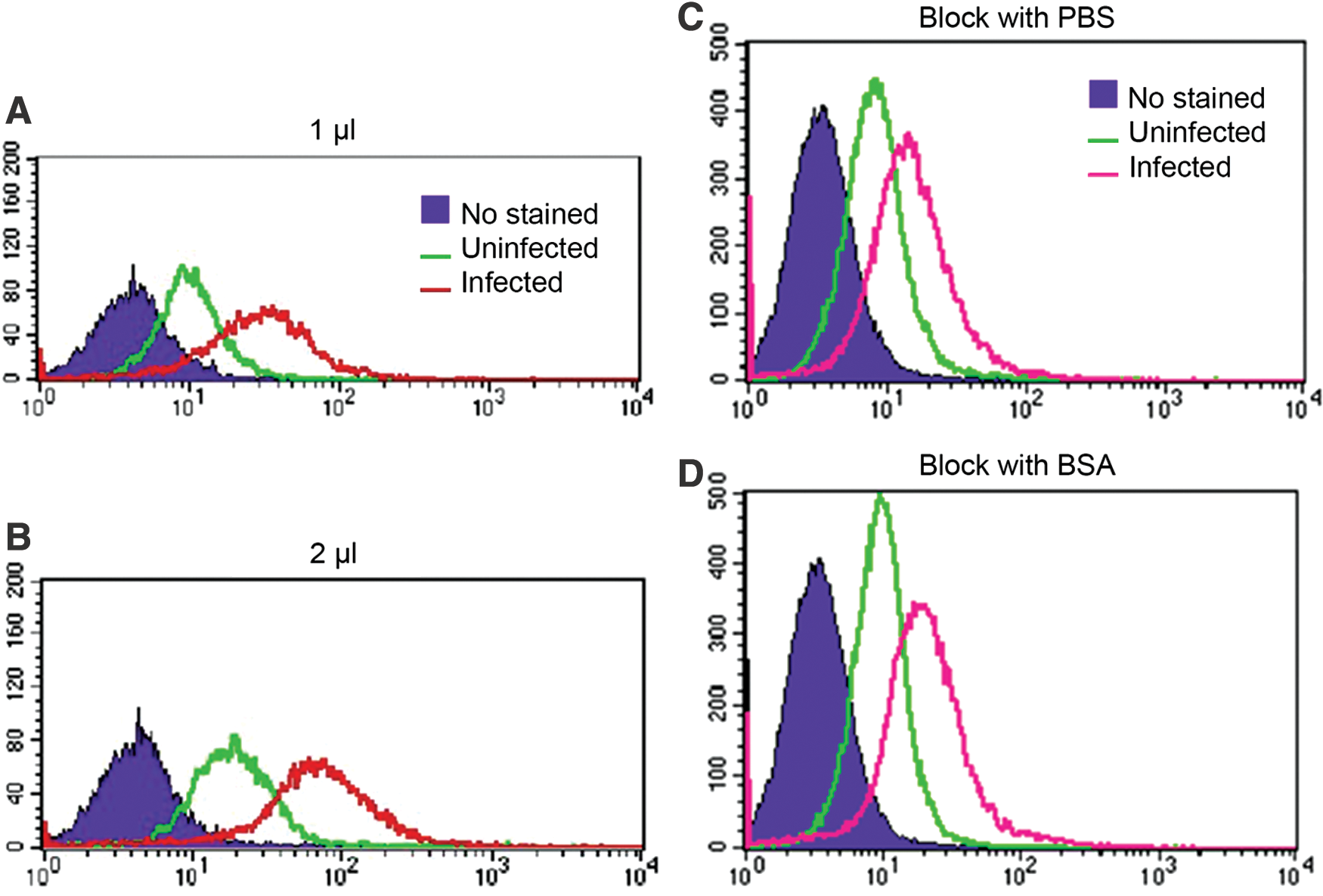
The proper staining amount and the necessity of blocking step in directly conjugated PE-anti-dengue virus antibody. Vero cells were infected with DENV at an MOI of 0 (uninfected) and 0.1 for 2 h and washed twice with PBS. The cultures were maintained in the maintenance medium for another 3 days before performing single fluorescence staining.
To increase the efficiency of the conjugated antibody, we reduced the amount to 1 μL and compared it with the previous 2 μL. Flow cytometry analysis showed that the peaks of both amount were similar (Fig. 1A, B). Thus, 1 μL of the directly conjugated antibody was efficiently enough, leading to the efficiency to stain more experiments per stock of the conjugated antibody.
Since the fluorescence intensity peaks of uninfected cells were higher than no-stained cells (Fig. 1A, B), the nonspecific binding might be the cause leading to the misinterpretation of actual infected cells. To ensure that there was no nonspecific binding to the noninfected cells, uninfected and DENV-infected Vero cells were blocked with 3% BSA and compared with PBS as a negative control before staining with the conjugated antibody. Flow cytometry analysis showed that the difference of uninfected and infected peaks were similar between blocked and nonblocked cells (Fig. 1C, D). Thus, the conjugated antibody did not require the blocking step from 3% BSA leading to reduced time used in the staining protocol.
Double simultaneous detection using FITC-anti-CD41a antibody and directly conjugated PE-anti-DENV antibody
To test the capability of directly conjugated PE-anti-DENV antibody for double-fluorescence staining, this conjugated antibody and FITC-anti-CD41a antibody were tested to simultaneously detect DENV and CD41a of DENV-infected MEG-01 cells. MEG-01 cells are well known to express CD41a on their cell surface (4) and are susceptible for DENV infection (3). Therefore, MEG-01 cells were used as a model for the testing. MEG-01 cells were infected with DENV at an MOI of 1. At 7 dpi, the double-fluorescence staining was applied to both uninfected and DENV-infected cells. The cells were fixed, permeabilized, and stained with both FITC-anti-CD41a mAb and directly conjugated PE-anti-DENV mAb, similar to the staining protocol used in Vero cells. Flow cytometry analysis of the uninfected cells showed dim CD41a (Fig. 2B). Similarly, the analysis of the infected cells showed bright intracellular DENV, but dim CD41a (Fig. 2C).
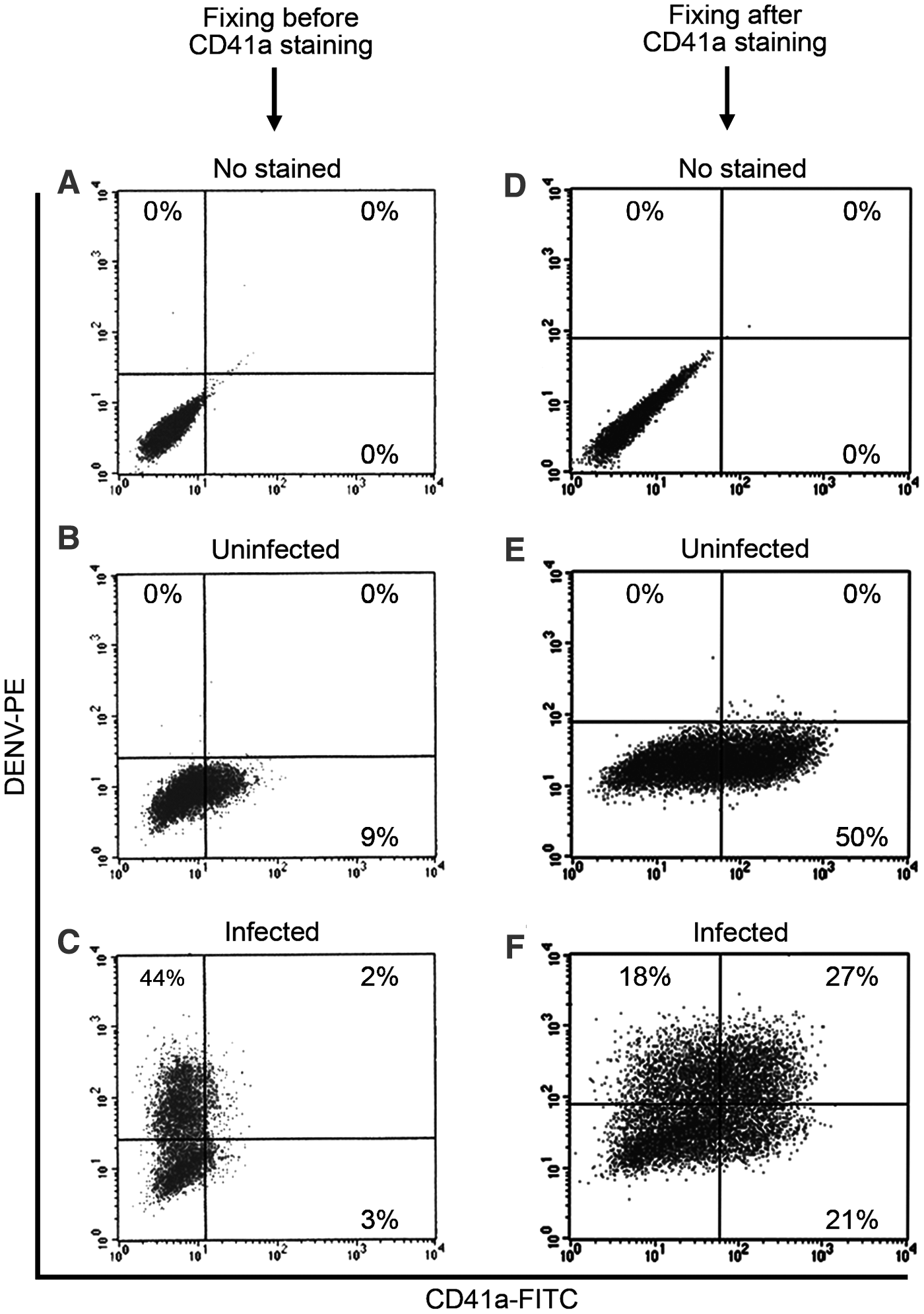
Comparing between fixing before and fixing after CD41a staining followed by dengue virus staining. MEG-01 cells were infected with DENV at an MOI of 0 (uninfected) and 1 for 2 h and washed twice with PBS. The cultures were maintained in the maintenance medium for another 7 days before performing
We next adjusted CD41a staining protocol because CD41a must be brightly expressed on cell surface of MEG-01 cells (4). It is a specific surface marker for human megakaryocytes to specify MEG-01 cells as a human megakaryoblastic cell line (7). Therefore, the protocol must have been improved until the signal on the cell surface was clearly detected by flow cytometry. We hypothesized that 4% paraformaldehyde might destroy CD41a. Therefore, we adjusted the protocol to first stain CD41a before the cells were fixed with 4% paraformaldehyde and permeabilized. Flow cytometry analysis showed that CD41a on cell surface was detected significantly brighter (Fig. 2E) when compared to the previous protocol (Fig. 2B, C). Moreover, intracellular DENV were clearly detected in both CD41a− (18%) and CD41a+ cells (27%) (Fig. 2F). Thus, CD41a must be immunostained before cells are fixed and permeabilized.
We further improved the efficiency of DENV infection by reducing the MOI from 1 to 0.5. MEG-01 cells were infected at an MOI of 1 and 0.5. The infected cells were stained at 7 dpi. Flow cytometry analysis showed similar results of the percentage of infectivity comparing between MOI of 1 (15 + 36 = 51%) and 0.5 (14 + 39 = 53%) (Fig. 3C, D). Thus, MOI of 0.5 was efficiently enough for DENV infection leading to the efficiency to infect more experiments per stock of DENV.
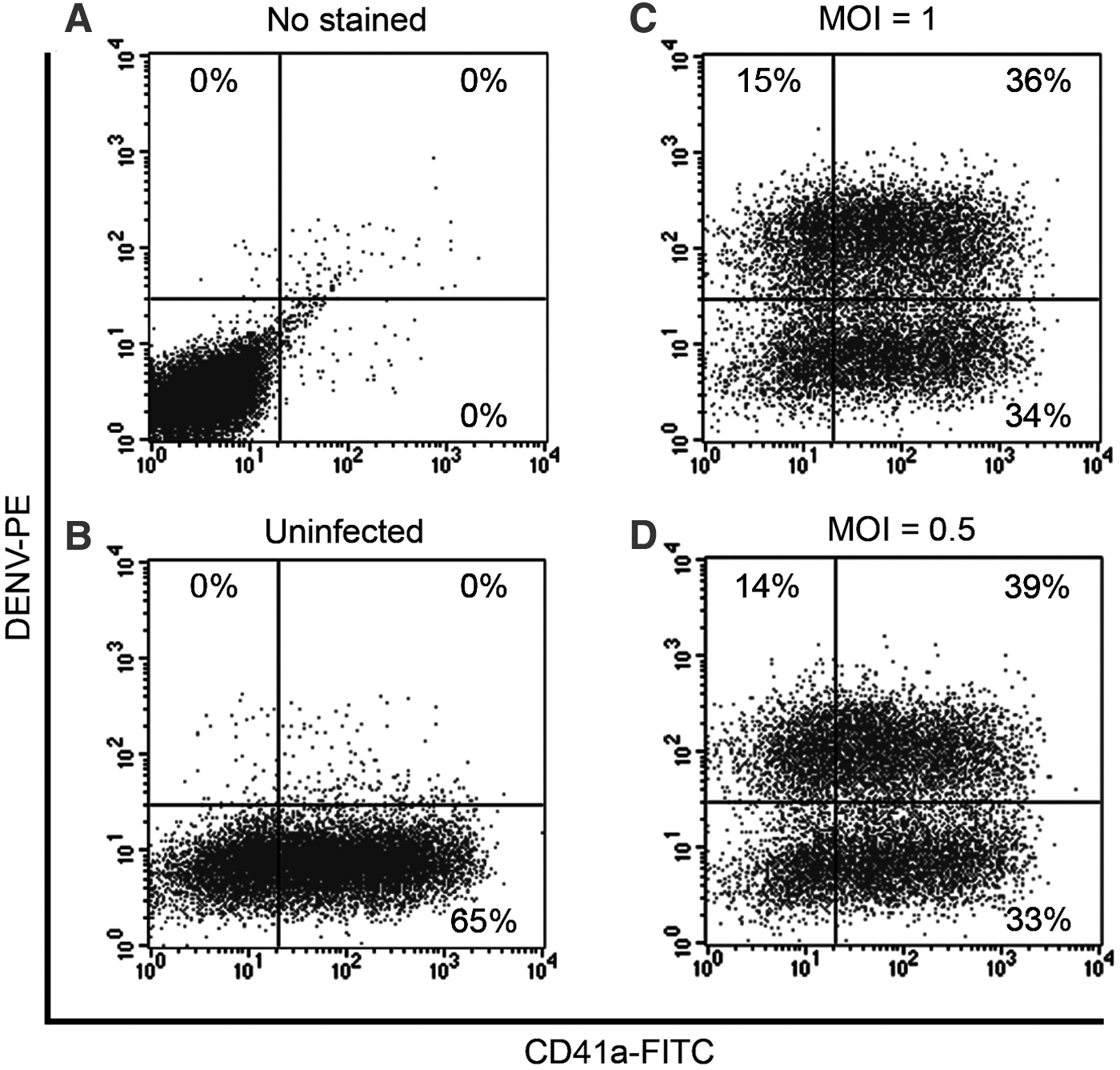
Monitoring the efficiency of dengue virus infection by directly conjugated PE-anti-dengue virus antibody. MEG-01 cells were infected at an MOI of
We further tested the capability of the conjugated antibody to identify the kinetics of DENV. MEG-01 cells were infected with DENV at an MOI of 0.5. The infected cells were collected at three different time points, including 2, 4, and 6 dpi. Flow cytometry analysis showed that DENV started being detected in both CD41a− and CD41a+ cells at 2 dpi (7 + 11 = 18%) (Fig. 4A). The analysis further showed that DENV was increasingly detected at 4 dpi (18 + 23 = 41.0%) (Fig. 4B). However, the detected DENV were not different at 6 dpi (14 + 26 = 40%) compared with 4 dpi (41.0%) (Fig. 4B, C). Thus, 4 dpi was enough for DENV to fully infect MEG-01 cells.
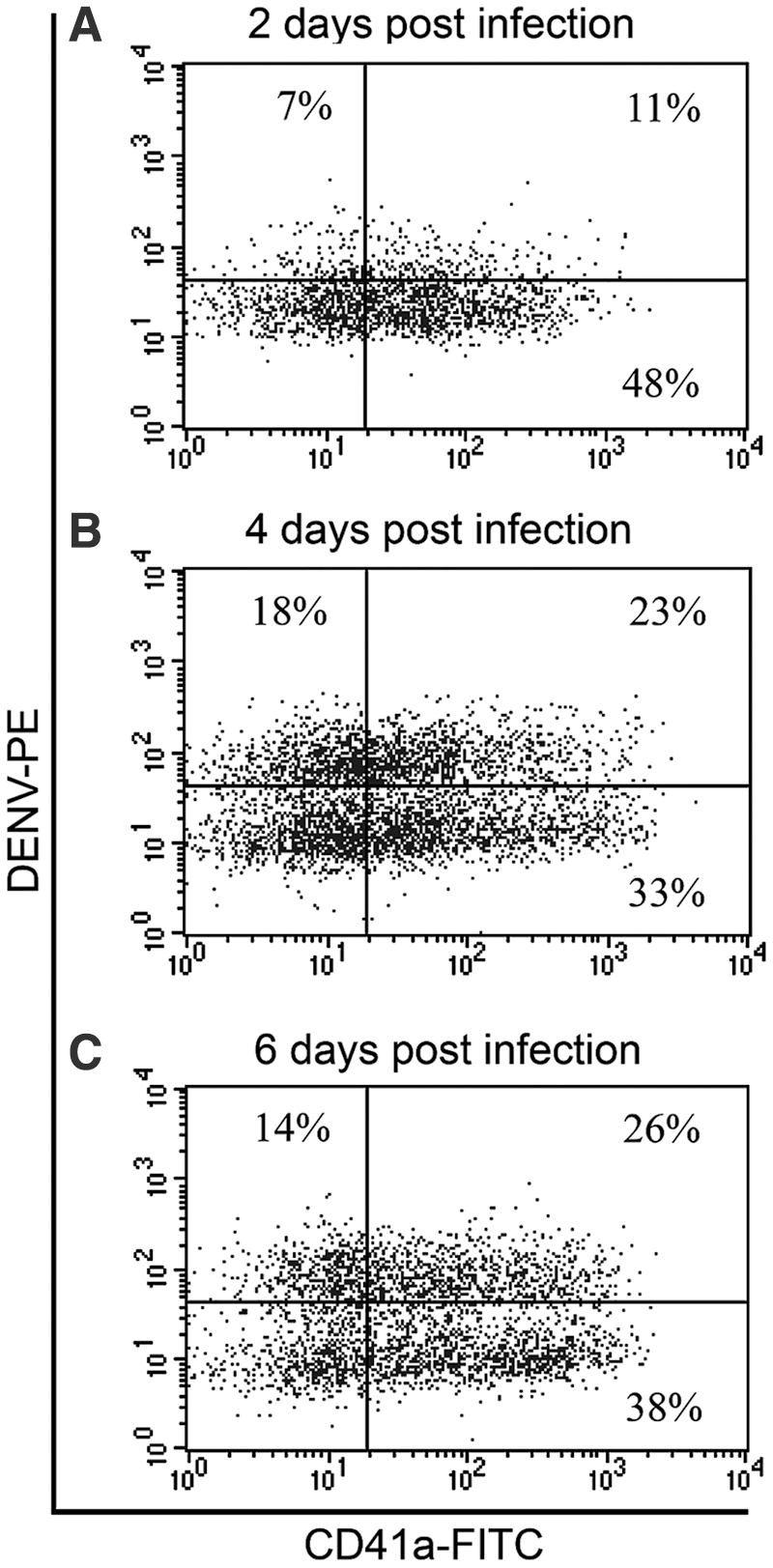
Kinetics of dengue virus infection by directly conjugated PE-anti-dengue virus antibody. MEG-01 cells were infected at an MOI of 0.5 for 2 h and washed twice with PBS. The cultures were maintained in the maintenance medium for another
Specifying the binding location of FITC-anti-CD41a antibody and directly conjugated PE-anti-DENV antibody
Flow cytometry can demonstrate the signal only as positive or negative. It cannot show where the signal localizes (Figs. 3C, D, 4A–C). First, we did immunostain CD41a before we permeabilized the cells. The antibody must have not been able to pass the cell membrane. Next, we did immunostain DENV after we permeabilized the cells. The antibody now can pass the permeabilized cell membrane into the cytoplasm. To demonstrate the localization, we analyzed those immunostained cells under fluorescence microscope. The cells were double stained with both antibodies at 4 dpi. For the MOI of 0, fluorescence microscope analysis showed that CD41a was detected superficially without any detected DENV. For the MOI of 0.5, the analysis showed three different detections as the following: first, CD41a appeared superficially without intracellular DENV (Fig. 5B; green arrow). Second, DENV appeared intracellularly without superficial CD41a (Fig. 5B; red arrow). Third, superficial CD41a and intracellular DENV appeared simultaneously (Fig. 5B; white arrows, 5C, D). DENV were detected in both CD41a− and CD41a+ cells similarly to the flow cytometry analysis (Figs. 3C, D, 4A–C). Thus, FITC-anti-CD41a antibody bound to CD41a at a surface level, whereas PE-anti-DENV antibody bound to DENV at an intracellular level.
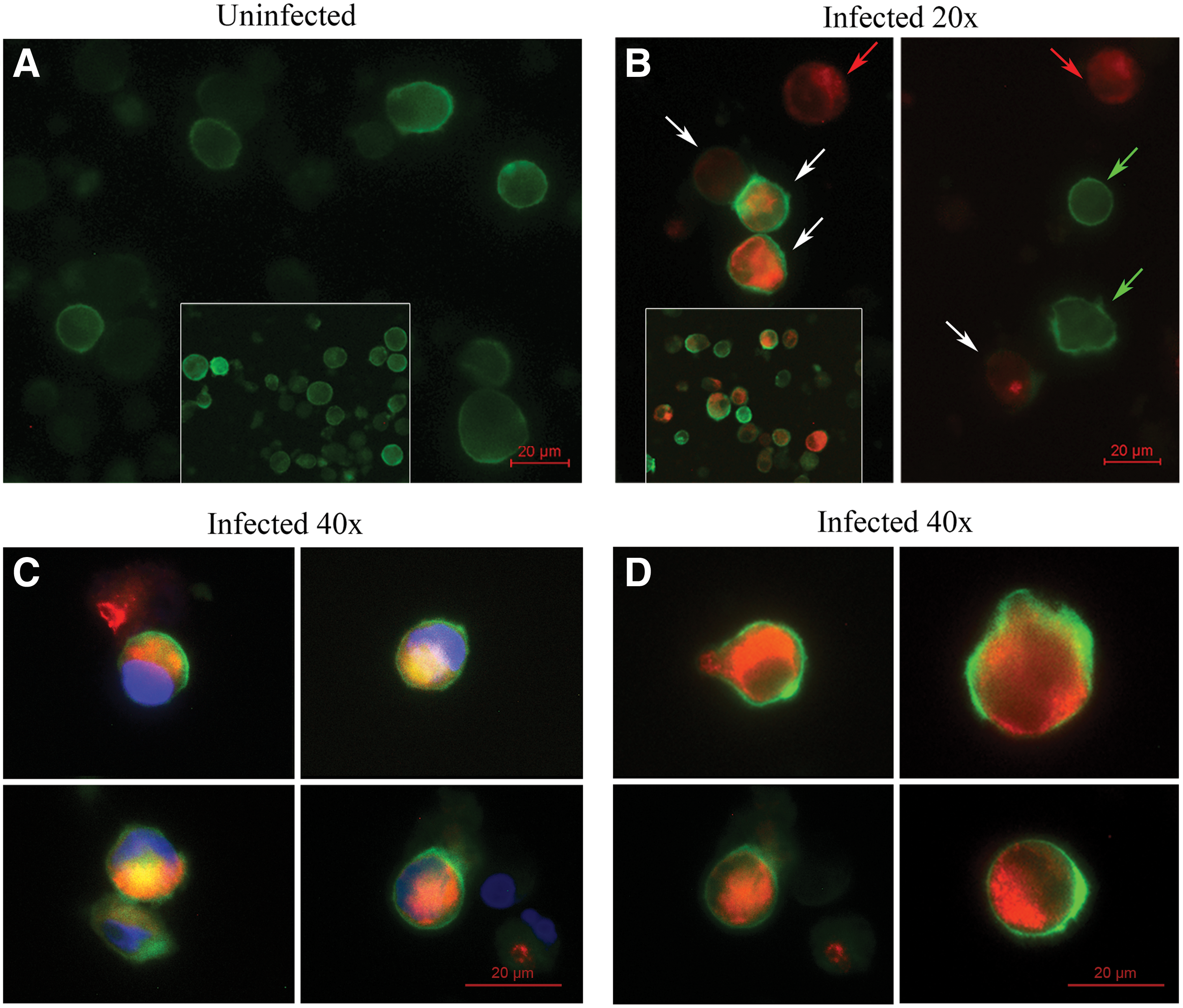
The binding location of FITC-anti-CD41a antibody and directly conjugated PE-anti-dengue virus antibody. MEG-01 cells were infected with DENV at an MOI of 0 (uninfected) and 0.5 for 2 h and washed twice with PBS. The cultures were maintained in the maintenance medium for another 4 days before performing
Discussion
DENV infections of megakaryocytes, which produce platelets and the subsequent events, have been proposed to be a factor contributing to thrombocytopenia. Until now, the capability of megakaryocytes to be a host of DENV still remains unclear (11). To answer this question, a methodology to simultaneously detect intracellular DENV and surface marker of megakaryocyte is definitely required. The major obstacle to develop this method was that there was no directly conjugated fluorochrome anti-DENV mAb commercially available. This article put through this obstacle by directly conjugating an interesting fluorochrome to anti-DENV primary antibody through LYNX Rapid Conjugation Kits.
Due to the presence of a small fraction of megakaryocytes (0.05%) in all nucleated human bone marrow cells (14), MEG-01 cell (human megakaryoblastic leukemic cell line) was used instead of human megakaryocytes in many articles (9,12). Although many immortalized megakaryocytic cell lines were successfully established, MEG-01 cells display phenotypic properties more closely to megakaryocyte than other cell lines (8). In culture, MEG-01 cells have the ability to spontaneously produce platelet-like particles very similar to human platelets, especially the expression of specific markers (12). Although the infection of MEG-01 cells by DENV was previously reported using EM (3), there are many drawbacks of this technique, especially an inconvenience for detecting intracellular DENV and surface marker of megakaryocytes simultaneously.
Thus, immunofluorescence staining technique was chosen for overcoming the inconvenience. This technique in conjunction with flow cytometry analysis can detect more than 15 markers simultaneously with different emission spectra depending on a particular model of flow cytometry (2). However, one important obstacle of the current objective was that there was no commercially directly conjugated fluorochrome-anti-DENV antibody, whereas it was essentially needed for multiparameter analysis. Consequently, the attempts were made to directly conjugate a primary anti-DENV mAb with PE through commercially available LYNX Rapid Conjugation Kits. As the results of the efforts, a simple method for directly conjugating PE to the primary antibody has successfully been demonstrated herein.
Flow cytometry analysis is rapid, high-throughput, and quantitative. Moreover, flow cytometry is able to analyze numerous cell types simultaneously by using a combination of different specific fluorescent biomarkers (which do not cause any spectral overlap). The multiparameter analysis of double-stained cells give investigators an advantage to identify which cell type is infected, as well as to quantitate a number of virus-infected cells (1). By utilizing this advantage, this study demonstrated that DENV were able to infect both CD41a− and CD41a+ MEG-01 cells (Figs. 3 –5). It suggests that DENV could infect both CD41a− cells and CD41a+ cells in dengue patients. The infection of CD41a+ cells such as megakaryocytes and platelets could lead to the destruction of these cells, resulting in thrombocytopenia.
Notably, another advantage of double immunofluorescence staining is the fact that it can be analyzed by both flow cytometry and fluorescence microscopy. There are many disadvantages of fluorescence microscopy compared to flow cytometry as follows: (1) flow cytometry is more sensitive than fluorescence microscopy, at least when the latter relies on human eye for qualitative fluorescence detection. (2) The duration of observation in fluorescence microscopy is necessarily longer than in flow cytometry, thus possibly contributing to neglect a fraction of positive cells due to fluorescence bleaching. (3) Quantitative analysis using fluorescence microscopy is more difficult and labor-intensive. However, fluorescence microscopy analysis can show where the antibodies bind to cells, whereas flow cytometry analysis cannot. Green-CD41a was demonstrated more superficial than RED-DENV, suggesting that CD41a is on surface and DENV is in cytoplasm. (Fig. 5C, D). Therefore, this study used fluorescence microscopy analysis to prove that anti-CD41a mAb bound to CD41a at a surface level, whereas anti-DENV mAb bound to DENV at an intracellular level.
This study described the simple method for directly conjugating fluorochrome to a primary anti-DENV mAb to detect DENV infection in CD41a− and CD41a+ cells. Given a simple protocol to follow and obvious results shown in this study, this strategy can facilitate virologists for directly conjugating any virus-specific primary antibodies, which are not commercially available with fluorochrome, to study the infectivity in any surface marker-specific host through flow cytometry. Together, we were able to conclude that DENV interacted with both human gpIIb/IIIa− and gpIIb/IIIa+ cells.
Footnotes
Acknowledgments
The authors thank Watcharee Attatippaholkun for helpful discussion and editorial assistance, and Sansanee Noisakran and Pucharee Songprakhon for DENV type 2 (strain 16681).
This research project was funded by grants from the Thailand Research Fund (grant no. RTA 488–0007) and the Commission on Higher Education (grant no. CHE-RES-RG-49). Surapol Issaragrisil is a Senior Research Scholar of Thailand Research Fund.
Nattapol Attatippaholkun is a PhD candidate in Molecular Medicine and a MD-PhD Scholar of Medical Scholars Program at Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand. This work is submitted in partial fulfillment of the requirement for the PhD.
Authors' Contributions
Nattapol Attatippaholkun designed and executed the experiments, and wrote the article. Yaowalak U-Pratya, Panthipa Supraditaporn, and Chanchao Lorthongpanich suggested the experimental protocols. Kovit Pattanapanyasat provided the flow cytometry facility. Surapol Issaragrisil supervised and funded the study. All authors analyzed the results and reviewed the final article.
Author Disclosure Statement
No competing financial interests exist.