
Abstract
Influenza A virus (IAV) imposes a significant socioeconomic burden on humanity. Vaccination is effective in only 60% of individuals, even under optimal circumstances. The difficulty stems from the remarkable ability of IAV to evade existing immunity. IAV's error prone polymerase enables the rapid antigenic evolution of the two virion surface glycoproteins, neuraminidase and hemagglutinin (HA). Since the most potent antibodies (Abs) at neutralizing viral infectivity are directed the head of the HA, amino acid substitutions in this region enable IAV to evade Ab-based immunity. Here, we review recent progress in understanding how immunodominance, the tendency of the immune system to respond to foreign immunogens in a hierarchical manner, shapes IAV evolution.
The Challenge: Vaccine-Resistant Human Viruses
M
A number of viruses that have evaded the standard vaccine strategies remain as significant human pathogens. These includes two viruses, human immunodeficiency virus (HIV) and influenza A virus (IAV), with very different replication strategies, but with a degree of antigenic variation in viral surface proteins sufficient to thwart standard vaccine strategies. For both HIV and IAV, robust neutralizing antibody (Ab) responses can be generated against various standard vaccine candidates, but ongoing viral evolution seriously hinders vaccine efficiency.
The elusiveness of IAV as a vaccine target has been known for 70 years (15), with the term “antigenic drift” being coined by 1965 (3). We now know that antigenic drift is based on the accumulation of amino acid substitutions in viral glycoproteins that gradually alter their antigenicity (36). Drift is mainly driven by selection of viruses that are resistant to neutralizing Abs (5), though antigenically relevant substitutions can be stochastically co-selected by other factors, including altered viral binding to host cell receptors (23).
Influenza A virions possess two surface proteins targeted by neutralizing Abs. The hemagglutinin (HA), a homotrimeric glycoprotein is present at about 400 spikes per virion (Fig. 1) (27,44,57). HA functions to attach virions to terminal sialic acids on cell surface glycoproteins and glycolipids and then to mediate fusion of viral and cellular membranes when virions are exposed to the acidic pH of internalizing endosomes. HA-specific Abs can neutralize virus by blocking attachment or membrane fusion (53). Such Abs are typically specific for the globular domain of HA, the location of the sialic acid binding site. The globular domain is hypervariable, exhibiting an average of 1.5 substitutions each year (6). Substitutions strongly tend to localize in distinct antigenic sites (Fig. 2), originally delineated by selecting neutralization escape mutants with monoclonal Abs (mAbs). Multiple mutations are needed to abrogate binding of Abs binding nonoverlapping epitopes, thereby defining distinct sites (68). This latter point is often underappreciated, as it shows first, that the effects of amino acid substitutions on antigenicity are limited to their immediate surroundings, and second, that the definition of antigenic sites is not completely arbitrary by nonstructural approaches.
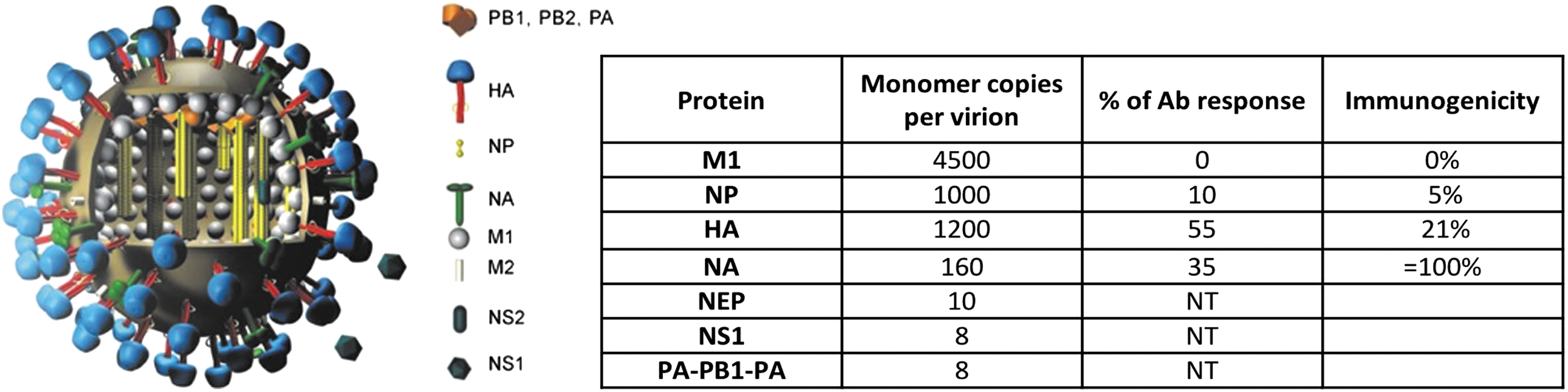
Pattern of ID to IAV virion. Cartoon of virus showing the structural proteins as indicated. Table shows the relative abundance of surface and internal proteins (27). The relative ID of serum Abs in response to infection and immunization is not linearly related to the copy number of virion of proteins. Ab, antibody; HA, hemagglutinin; IAV, influenza A virus; ID, immunodominance; NA, neuraminidase.
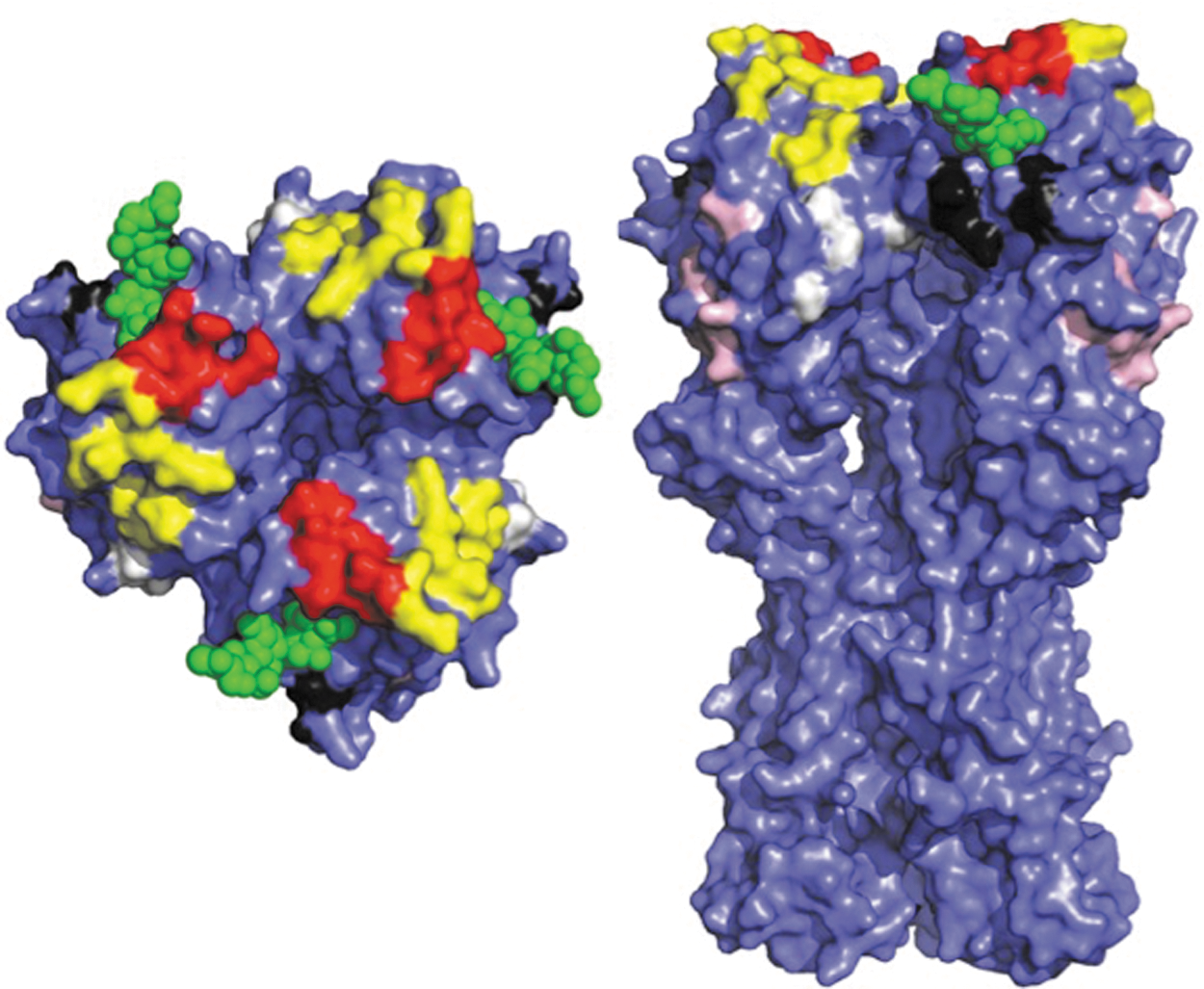
Defined antigenic sites on PR8 HA. Sites defined by mAb escape mutant selection (7) are shown on the HA structure (PDB 1RU7) (18). Each monomer in the native trimer is colored purple. Bound sialic acid is shown in green. Antigenic sites: Sa, yellow; Sb, red; Ca1, white; Ca2, black; Cb, pink. Sa and Sb sites are strain specific, while Ca and Cb sites are shared by some drifted variants. Left, trimer, as it would appear looking at virion from top. Right, side view of trimer. Note that the transmembrane domain is not present in the solved structure.
The other viral glycoprotein, neuraminidase (NA), a homo-tetrameric glycoprotein, has the opposite function of HA. It removes terminal sialic acids, thereby releasing HA-bound virions from infected cells, mucous, or other host virion-sequestering substances. NA is present on virions at ∼10% the level of HA (Fig. 1). Abs specific for NA reduce viral infectivity by preventing viral release from terminal sialic acid containing substances. Though attention is focused on the HA for vaccine failures, NA accumulates surface amino acid substitutions more rapidly in its globular domain (6), consistent with an important role for NA in evading human humoral immunity (12). NA functions in close conjunction with HA (31); Ab selected substitutions in HA can select for epistatic substitutions in NA that alter its function and antigenicity (8,24,48). Further complicating matters, HA specific Abs can interfere with NA function on intact virions (33), consistent with the high ratio of HA to NA on virions (27) (Fig. 1) and the high density of spike proteins on virions (57).
Why Does IAV Drift?
While considerable progress has been made in understanding the interaction between IAV and Abs, the fundamental paradox of antigenic drift remains: why does IAV drift while other respiratory RNA viruses with similarly high mutation rates and antigenic escape frequencies (54,59,65) remain essentially antigenically static? This may ultimately be related to a high intrinsic plasticity of HA and NA in accepting multiple mutations relative to other viral glycoproteins (17,22). But even if true, this begs the question of why other viruses couldn't evolve similarly plastic proteins.
Given the presence of five independently mutable antigenic sites on the HA that bind neutralizing Abs (7), the immune system would seem to have a stable target, if even pressure could be exerted by Abs against all of the sites. Even with the high frequency of individual antigenic escape mutants (10−5) (58,68), the frequency of a five site spontaneous escape mutant would be 10−25. This is a considerable amount of virus: 1025 virions would weigh ∼107 kg, filling four Olympic swimming pools, and roughly represent 104 times the total number of infectious virions produced by the human population yearly [assuming 10% infection rate and 1012 infectious virions produced per human per year (51)]. Since more than one amino acid substitution is required to completely change each site (8), at least 10 changes in the globular domain are required to completely change antigenicity, meaning it would take on the order of a billion years to randomly generate a complete escape mutant in the human infected population!
Obviously, then, the entire human infected population does not apply even selective pressure against all antigenic sites. Indeed, shortly after it became possible to generate single substitution escape mutants with mAbs, it was reported that such mutants could escape polyclonal antisera from animals and humans (47,63), providing early evidence human immune responses can be highly skewed toward single antigenic sites. Two recent reports (25,40) show that a substantial fraction of individuals (up to 42% in some age groups) focus their immune response to the Sa antigenic site on pandemic H1 HAs to the detriment of vaccine induced immunity, as a natural drift amino acid substitution in this site greatly reduced serum potency in standard functional assays.
These findings (and many others in the 30-year literature following the molecular characterization of antigenic drift documenting large effects of single point mutations on antigenicity) point to the importance of understanding the immunodominance (ID) of B cell responses to IAV infection and vaccination.
Ab Immunodominance
“Immunodominance” was coined in 1966 to describe antibacterial Ab responses heavily biased to certain bacterial structural components (43). ID describes the strong tendency of the immune response to respond to complex antigens in a hierarchical manner, with higher ranking, “immunodominant” antigens potentially suppressing (“immunodominating”) responses to “subdominant” antigens.
With the advent of mAbs in the 1970s, ID studies were extended to IAV and other viruses by determining the frequency of hybridomas synthesizing Abs specific for antigenic sites defined by the mAbs (55). Ironically, while the B cell ID field was poised for a great leap forward with hybridoma analysis, mAb-created escape mutant panels, and mAb-Fab fragments with defined epitope specificity for use in serum competition studies (62), interest in Abs waned, as T cells became the focus of viral immunology.
Consequently, antiviral T cell ID was extensively characterized over the next three decades, particularly CD8+ T cells responses. The peptide-based nature of T cell receptor recognition facilitated quantitating T cell antiviral responses, which was further abetted by the development of MHC-peptide tetramers to directly enumerate T cells by flow cytometry (1). While knowledge of T cell ID is certainly far from complete (much less is known about CD4+ T cell ID), many of the major features of and factors contributing to CD8+ T cell ID have been delineated (66).
Although T cell-based IAV vaccines have never been tested in large clinical trials, the presence of robust numbers of memory T cells (1/250 of blood CD8+ T cells in most humans (21), and their apparent inability to provide protection (although, again, this has not been carefully correlated in large populations) returned focus on Ab-based vaccines. While anti-IAV Ab responses are technically easier to measure than T cell responses by standard binding and functional assays, it is more difficult to establish their epitope specificity.
With few known exceptions, Abs that bind to native to IAV HA and NA recognize discontinuous epitopes that require intricate protein folding (35). This rules out using synthetic peptides or peptide fragments generated biochemically or by genetic display technology to determine epitope specificity of most biologically relevant Abs. Quantitating polyclonal Ab (pAb) responses is further confounded by the problem of Ab affinity for the immunogen/antigen. Signal magnitude measured in simple binding assays and more complex functional assays is governed by the product of Ab concentration and avidity for antigen (note that in standard immunological terminology, affinity refers to monovalent Ab binding to antigen, while avidity refers to multivalent binding, as occurs with immunoglobulin (Ig) binding to multivalent antigens such as viruses). True avidity measurements of antiviral pAbs are essentially limited to the remarkable body of studies from Fazekas de St. Groth from 50 years ago (13). The immune system has gone to great lengths to evolve the process for Ab class switching, somatic hypermutation, and affinity maturation, yet we know little about the relationship between Ab avidity and antiviral functional activity, and the functional consequences of class switching on Ab direct antiviral functional activities.
pAb responses are also complicated by the presence of dozens (52) to thousands (55) of species that simultaneously compete for binding to their cognate antigen while exerting allosteric affects that can increase the affinity of noncompeting Abs (41). On top of this, Abs can interact with serum proteins (e.g., C1Q) that modulate Ab function by increasing steric interference effects on target antigens (45), and also enable interaction with innate immune cells that exert complex antiviral activities by phagocytosing viruses and by secreting cytokines and cytotoxic molecules when they encounter virus-infected cells (38). The latter is also achieved when Abs interact with innate cells (natural killer cells, macrophages, granulocytes, mast cells) via Fc receptors (9,10). Ab-guided-innate cells seem likely to play a much larger role in IAV protection than is currently appreciated (28).
Fundamentals of Anti-IAV Ab ID
To better understand and possibly even predict IAV antigenic drift with the goal of improving vaccines, it is essential to better understand B cell and Ab ID. This entails defining the ID hierarchy of Ab responses at the level of proteins, antigenic sites, and epitopes, and then delineating the underlying cellular and molecular mechanisms during naïve and memory responses.
To start, we addressed the simple, obvious, and yet unexplored question of the relationship between virion protein abundance and Ab ID (2). We gauged ID by simple enzyme-linked immunosorbent assay (ELISA) using glycoproteins purified from H3/H1 reassorted viruses and purified internal proteins, to measure Ab responses to M1, NP, HA, and NA, the most abundant viral structural proteins. Although the amount of antigen bound to ELISA plates varies, the titer, that is, the serum dilution to achieve half maximal (or any arbitrary fractional) binding, should be independent of bound antigen concentration based on first order mass action binding. We found that ∼2/3 of the serum responses of mice following immunization with inactivated IAV focuses on the HA, with the remaining Abs recognizing NA, and to a lesser extent nucleoprotein (NP). Notably, there is a poor correlation of ID with virion protein abundance. M1 did not induce a measurable Ab response, and NP ranked below NA, which on a molar basis, is the most immunogenic protein under these immunization conditions (Fig. 1).
Most remarkably, lampreys recapitulated the mouse ID Ab response hierarchy to IAV, despite having evolved a completely different type of immune cell Ab receptor, termed a variable lymphocyte receptor (VLR) (49). As with mice, guinea pigs, and chickens, lamprey Abs recognized the five principal antigenic sites on the HA head. We inferred this by using a panel of escape viruses that we had sequentially evolved in vitro using a panel of 12 neutralizing head specific mAbs (8). In all species tested, we failed to detect primary antistem Ab responses, demonstrating its low status in the ID hierarchy.
These findings suggest that the rules governing B cell ID are largely conserved between organisms, even those with a completely different basis of recognizing immunogens. This points to physiochemical features of complex antigens as the governing principal in their immunogenicity. Pragmatically, this is good news, as it supports the use of a diverse range of animal models in designing human vaccines for IAV and other viruses.
This does not preclude the possibility of species-associated differences in immunogenicity: indeed, it is likely that VLRs will select a distinct, though clearly overlapping repertoire of escape mutants based on differences in the nature of the VLR and Ig combining sites. Further, structural differences between jawed vertebrate Igs [including the lack of light chains in camelid and shark Abs (32), extended complementary determining regions domains in cow Abs (61)] are almost certain to result in distinct, if overlapping epitope specificities. This can best be functionally assessed by detailed characterization of escape mutants selected with mAbs and physically, by structural definition of mAb-HA interactions (19).
Ab ID at the Level of Individual Antigenic Sites
To increase the resolution of pAb ID analysis to the level of individual antigenic sites we created a panel of “Δ4”sequential escape mutants designed to simultaneously ablate four of the five defined PR8 HA globular domain antigenic sites (Fig. 2) (4). This was the model antigen for Walter Gerhard's pioneering work of studying the diversity of Ab responses using mAbs (67), and is still probably the most painstakingly defined antigen, due to the large number of mAbs analyzed and the self-reporting nature of viral escape mutants based on RNA sequencing of epitope residues (7).
Using the Δ4 panel viruses as ELISA antigens, we could quantitate pAb responses, and Ab secreting cells by ELISPOT. By creating an equivalent panel of recombinant HAs suitable for flow cytometry by mutating the sialic binding site to prevent nonspecific binding to B cells and adding a biotin labeling site (64), we could in parallel quantitate B cell subsets by flow cytometry (though flow analysis was limited to germinal center B cells, since other primary B cells express little or no cell surface Ig, preventing their detection by live-cell flow cytometry). This analysis led to a number of findings that begin to delineate ID in mouse anti-HA Ab responses.
B cell and Ab ID is well ordered but dynamic and under genetic control
After infecting C57BL/6J mice intranasally with the mouse adapted PR8 strain, we could first detect B cell responses on D7 in the form of Ab-secreting extrafollicular B cells, predominantly focused on Cb responses. The first germinal center (GC) B cell responses, detected on D14, also were dominated by Cb specific cells. This is consistent with prior studies demonstrating the dominance of Cb Abs in early primary responses (30). By day 21, Sa-specific B cells were detected, to be joined a week later by GC B cells specific for each of the sites. Parallel measurements of serum Ab responses revealed a generally good (but imperfect) correlation between serum Ab responses and GC B cell frequencies, consistent with GC B cells as the principal source of plasmablasts/plasma cells believed to be the primary producers of serum Abs.
Importantly, our findings, in conjunction with two other recent studies (34,56), suggest distinct rules for GC B cell interclonal versus intraclonal competition. Responses to haptens and other single-epitope antigens are mainly driven by intraclonal competition for highest affinity B cell. By contrast, in responses to HA and other multi-epitope antigens a key element is interclonal competition of B cells of different specificities. It appears that independent GCs exhibit different affinity thresholds, favoring increased clonal diversity as the response matures.
The greatest discrepancy in GC B cell numbers and serum Ab responses occurred with Sb-specific responses. Sb-specific Abs were nearly co-dominant in serum at day 21 while Sb-specific B cells were not detected in GCs. The relationship between B cell numbers and serum responses, measured by ELISA area under curve values would be heavily skewed if there were large antigenic site-specific difference in the average Ab avidities. By staining GC B cells with graded amounts of labeled HA, we could estimate their average avidity (16), revealing a threefold difference between the lowest (Ca2) and highest affinity (Cb) B cells. So, affinity differences do not explain the disconnect in Sb responses, which could be a technical issue, or indicative of an extra-follicular source of Sb-specific plasmablasts/plasma cells.
To examine the influence of mouse strain on the ID hierarchy, we repeated the experiment with BALB/c mice. Like C57BL/6J mice, the BALB/c hierarchy was dynamic and reproducible, but differed in site specificity, with Sb-specific responses dominating throughout the response, and Sa-specific Abs being higher. Experiments are in progress using collaborative cross mice (14) to explore the genetic contribution to these differences, and to what extent an ID phenotype maps to Ig- versus non-Ig genes.
The initial ID hierarchy is not dependent on CD4 cells
Depleting CD4 cells profoundly reduced Ab responses to intraperitoneal immunization as expected (37,46), accompanied with lack of GC formation, class switching, and rapid decline of Ab titers. It had a surprisingly minor effect, however, on the serum Ab ID hierarchy, with the exception that Sb-specific responses were selectively suppressed relative to other antigenic sites. This suggests that the initial ID hierarchy is largely established by the B cell repertoire, though Sb-specific B cell expansion is particularly dependent on T cell help.
ID hierarchy is altered by immunization conditions
Vaccination, particularly intramuscular vaccination with semi-purified HA, the standard in most countries, clearly differs from natural infection. How does this affect the Ab repertoire? C57BL/6J mice immunized by intraperitoneal or intramuscular injection of intact inactivated virus demonstrated a greatly altered ID hierarchy, now dominated by Sa and Sb Abs, well correlated with the splenic GC response (Sb was still under-represented in GC B cells). In further contrast to intranasal infection, serum Ab hierarchies were relatively static between days 14 and 28.
GC B cell frequencies in the mediastinal lymph node and spleen differed in a site-specific manner between mice infected intranasally versus those immunized intraperitoneally, demonstrating that lymphoid organ-specific differences in B cell repertoires could not account for immunization-based differences in GC B cell ID hierarchy. Rather, it appears that the form of antigen presented to B cells, in conjunction with antigen availability, governs the ID hierarchy, particularly since as discussed above, the initial ID hierarchy is largely independent of T cell help.
The HA stem is poorly immunogenic in primary responses
In all conditions tested, stem specific-Ab titers were lower, and usually much lower than head-specific titers, though intramuscular immunization resulted in a more balanced early response. Antistem responses typically demonstrated very little increase with time (day 14–28), strikingly so for intramuscular immunization, where they barely budged. As an important exception, antistem titers increased sixfold following intraperitoneal immunization, and still increased twofold in the absence of CD4+ T cells, while antihead Abs declined slightly. This suggests that stem- and head-specific B cells are governed by different rules, and emphasizes that the route of immunization can greatly affect the ID hierarchy.
Abs modulate the ID hierarchy
A critical feature of B cell responses is that secreted Abs compete for binding with the B cell surface Igs. This is particularly important for IAV vaccination, since all but the youngest children have either been infected or vaccinated with IAV. We found that site-specific Abs administered either passively as a mAb Fab fragment (to minimize competition and avoid Fc mediated effects) or induced actively by infection with one of the Δ4 viruses (to better mimic human responses), selectively suppressed primary responses to the cognate antigenic site when mice were challenged with wt virus. This is consistent with mathematical modeling of anti-IAV Ab responses (69).
The Fab-mediated blockade was highly specific for the cognate Sb antigenic site. Despite suppression of the immunodominant Sb-response on d28, responses to other sites did not increase, indicating a lack of “immunodomination,” a phenomenon often observed in CD8+ T cell responses, in which dominant clones suppress lower hierarchy clones. The SbΔ4 virus immunization induced Ab-mediated blockade was also specific for the cognate site (Sb). This is unexpected, given that mAbs to Sb have been reported to bind competitively with mAbs specific Sa, Ca, or Cb in an ELISA type assay (42). How the steric effects could differ in vivo is puzzling, and suggests that other factors are in play.
Site-specific Ab activity
We could also use the Δ4 virus panel to measure the specific activity ( = functional titer/ELISA titer) of pAbs directed to each of the antigenic sites. Decades ago, the Gerhard lab had measured the specific activity of mAbs in hemagglutination inhibition (HI) and virus neutralization (VN) assay (U/mg). These varied based on proximity to the receptor binding site, with Sa-specific mAbs being 260-fold more efficient than Cb Abs in VN assays, and 60-fold more efficient in HI assays (note that this analysis is based on molarity, without taking Ab avidity into account).
Our findings with mouse pAbs induced by intranasal infection differed somewhat from the mAb panel, with Sa pAbs being the most efficient at both VN and HI (HI or VN titer/ELISA area under curve value), followed by Sb (HI), Ca1 = Ca2, and Cb being the least efficient. The efficiency difference between Sa and Cb pAbs was much less in VN than HI assays, consistent with the ability of many, if not most globular domain-specific mAbs, to neutralize postvirion attachment to host cells (11).
Strikingly, Sb-pAbs demonstrate very low efficiency at HI, while being second most efficient at VN. This is perplexing, since these Abs are defined by escape mutants selected by Sb mAbs that give HI with high efficiency. One possible explanation is that a class of Sb Abs not present in the characterized mAb panel, bind to the Sb antigenic site at an “atypical” angle, that is, away from the sialic acid binding site toward the neighboring protomers. While plausible, it would still mean that the bulk of Sb-specific mAbs would have to bind this way, and consequently that the hybridoma panel is heavily biased. Indeed, this highlights our lack of fundamental understanding of specificity-differences between distinct B cell compartments (GC B cells, plasmablasts/plasma cells, long-lived plasma cells, and memory B cells) and serum Abs.
Given the likely importance of Fc mediated protection implied by results from the mouse model (9,10,26,28), it is of obvious importance to extend these findings to activation of Fc bearing cells and Fc-mediated viral phagocytosis. Recent findings demonstrate the complexity of activating Fc receptors on NK cells, where Abs work both synergistically and antagonistically based on their binding domains on the HA (20,39). Potent HI Abs fail to induce antibody dependent cellular cytotoxicity (ADCC), but also block ADCC mediated by stem-specific Abs. What are properties of Sb-specific Abs that lack HI activity? What is the net effect of all of this complexity, and can the immune response eventually be tailored to optimize desired activities?
Whither ID?
While Abs have been a central topic of immunology since their discovery in 1890 (60), far less is known about Ab ID than T cell ID, despite the nearly 70-year head start. Although it is clear from decades of studies of myriad antigens that Ab responses focus on particular proteins or protein domains, there has been little systematic study of this phenomenon. This is unfortunate, since understanding ID requires better understanding of a multitude critical mechanistic components of the B cell response: antigen delivery to B cells in various physical locales, delivery of proper activation and survival signals by T helper cells and probably other immune cells, the base Ig repertoire to bind to a given antigenic site and capacity for honing of the repertoire by somatic hypermutation. Further, it is clear that a better understanding of Ab ID is required to improve vaccines to IAV and other higher hanging fruit pathogens, which have proven to be difficult vaccine targets.
We have limited this review to our recent work on IAV, but we hope that the reader will be able to extrapolate our strategies and findings to their virus/pathogen/antigen of interest. There are certain to be some important differences between different systems, but it is likely that there will be many more commonalities, and each system will offer its own unique advantages to generate findings that contribute to understanding the general rules of ID.
Footnotes
Acknowledgment
The authors are supported by the Division of Intramural Research, NIAID.
Author Disclosure Statement
No competing financial interests exist.