
Abstract
To understand the mechanism for inhibition of hepatitis B virus (HBV) infection is important. In this study, single-chain variable fragment (scFv) antibodies were generated and directed to the pre-S2 epitope of HBV surface antigen (HBsAg). These human scFvs were isolated from a person with history of HBV infection by phage display technology. An evaluation of panning efficiency revealed that the eluted phage titer was increased, indicating that specific clones were enriched after panning. Selected scFvs were characterized with the recombinant HBsAg through Western blotting and enzyme-linked immunosorbent assay to confirm the binding ability. Flow cytometry analysis and immunocytochemical staining revealed that one scFv, S17, could recognize endogenous HBsAg expressed on the HepG2215 cell membrane. Moreover, the binding affinity of scFv S17 to the pre-S2 epitope was determined to be 4.2 × 10−8 M. Two ion interactions were observed as the major driving forces for scFv S17 interacting with pre-S2 by performing a rational molecular docking analysis. This study provides insights into the structural basis to understand the interactions between an antibody and the pre-S2 epitope. The functional scFv format can potentially be used in future immunotherapeutic applications.
Introduction
T
Studies have confirmed that two specific regions, pre-S1 and pre-S2, which are present on the outer surface of the HBV envelope, play determining roles in HBV infection (6). Both regions are considered to be critical epitopes that can confer the broad protection offered by vaccination. Specifically, the pre-S1 region is responsible for attaching the virus particles to the host cell receptors (14) and the pre-S2 region contains two crucial factors, the pHSA site and pre-S2 glycan, which can recognize specific lectin on hepatocytes to assist the infection process (6). The pre-S2 mutants will affect the signaling of the infected cells and promote HBV-related tumorigenesis. Moreover, a transgenic model harboring a pre-S2 mutant was observed to induce nodular dysplasia and HCC (22). Mutations in these pre-S regions have resulted in the low expression of HBsAg (21), and different mutations or deletions have been reported to result in the reduction of binding affinity of monoclonal antibodies against HBsAg (16). These findings support the importance of the pre-S regions, not only can they induce neutralizing antibodies but also their variations may be associated with the progression of liver diseases.
Numerous studies have reported the feasibility of antibody-like molecules with a stabilized structure and specific binding ability for immunotherapeutic applications (19,20). Antibody drug conjugates (ADCs) are developed according to this concept, and many related clinical trials are ongoing (19). Recently, Krebs et al. used a chimeric antigen receptor T cell (CAR-T) system to provide immunotherapy for clinical HBV infection (10). In addition to the neutralizing effect of a specific binder (e.g., single-chain variable fragment [scFv]) itself, the binder can function as a missile, delivering an attached payload to the target site. Understanding the antigen–antibody interaction will facilitate the performance of antibody (scFv) engineering. In this study, we explored the interaction between the isolated scFv S17 and the pre-S2 epitope. We believe that our findings will be valuable in assisting the development of S17 for immunotherapeutic applications in the future.
Materials and Methods
Phage-displaying human scFv library and panning
To generate a specific anti-HBV pre-S2 epitope antibody, a phage-displaying scFv library was constructed from a person with history of HBV infection. The collection of human specimen in this study had been approved by the Institutional Review Board of Taipei Medical University before study initiation. Briefly, peripheral blood mononuclear cells (PBMCs) were freshly harvested from the blood for homogenization. Total RNA was extracted by using the RNeasy kit according to the manufacturer's instruction (Qiagen), and was reverse-transcribed into complementary DNA (cDNA) by the SuperScript III reverse transcriptase kit (Invitrogen). Furthermore, heavy and light chain variable (VH and VL, respectively) regions were amplified through polymerase chain reaction (PCR) by using specific antibody primers designed for humans. Then, two types of fragments were purified and subjected to a second round of PCR with a linker to form full-length scFvs, which were further digested with SfiI restriction enzyme (NEB Biolab) and cloned into the pComb3X phagemid vector (1). The recombinant library DNA was transformed into Escherichia coli ER2738 and rescued using VCS-M13 helper phage (Stratagene) for scFv display on the phage surface. Furthermore, the phage library was precipitated with 4% polyethylglycol 8000 and 3% NaCl (w/v), resuspended in phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA), and stored at 4°C.
The pre-S2 epitope peptide NSTTFHQALLDPRVRGLYFPAGG was designed from amino acids 123 to 145 of the HBsAg (GenBank Accession AAN03492.1). The peptide was synthesized, and a biotin molecule was conjugated at the C-terminal (Kelowna, Inc.). The binding clones in the prepared library were enriched through panning, using methods described elsewhere (20,21).
ScFv expression and purification
After panning, total library DNA was purified by the EasyPrep Plasmid Extraction kit (BioTools, Inc.) and transformed into E. coli TOP10F′ (a nonsuppressor strain; Invitrogen) for scFv expression. Clones were grown in 2YT medium at 37°C until the OD600 reached 0.6. After inducting with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 6 h, the bacterial cell pellet was resuspended in binding buffer (500 mM NaCl, 20 mM Tris–Cl, and 20 mM Imidazol; pH 7.4) and lysed by three cycles of freezing (−80°C) and thawing (37°C), and sonicated on ice till homogeneous. After centrifugation, the cellular lysate was loaded onto a Ni2+-charged resin column for His-tagged scFv purification according to the manufacturer's protocol (Amersham Biosciences).
Western blotting and enzyme-linked immunosorbent assay
To detect the binding reactivity of the selected scFvs, which were expressed as His and HA-tagged proteins, recombinant HBsAg were transferred onto nitrocellulose membranes (GE Healthcare Life Sciences) after sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The membranes were blocked with 5% skim milk, incubated with purified scFvs at room temperature for 2 h, and gently shaken. Next, the membranes were detected with mouse anti-HA antibodies and developed with horseradish peroxidase (HRP)-conjugated donkey anti-mouse immunoglobulin G (IgG) antibodies (Jackson ImmunoResearch Laboratories).
For enzyme-linked immunosorbent assay (ELISA), a microtiter plate was coated with recombinant HBsAg (0.5 μg/well) at 4°C overnight. After blocking with 5% skim milk, the expressed scFvs or scFv-expressing phages were added to the wells in duplicates and incubated for 1 h at room temperature. After washing with PBST (PBS with Tween 20), the scFv binders were detected as described above and the scFv-expressing phages were detected by using HRP-conjugated mouse anti-M13 phage antibodies (GE Healthcare Life Sciences). Finally, the 3,5,5-tetramethubezidine dihydrochloride substrate (TMB) was added for signal development. The reaction was stopped by adding 1 N HCl, and the absorbance was measured at 450 nm.
Sequence analysis
To sequence the interested scFv clones, a primer ompseq (5′-AAGACAGCTATCGCGATTGCAGTG-3′) complementary to the outer membrane protein A (ompA) signal sequence in front of the VL region was used. International ImMunoGeneTics information system/V-QUEry and STandardization (
Flow cytometry analysis and immunocytochemical staining
Hep3B cells were cultured in MEM supplemented with 10% fetal bovine serum. HepG2215 cells were cultured in MEM supplemented with 10% fetal bovine serum, 2 mmol/
HepG2215 cells were individually seeded on a cover glass and fixed by incubating with freshly prepared 4% paraformaldehyde (Sigma) on ice for 10 min. The cells were dehydrated with a sequential treatment of 70%, 95%, and 99% methanol and rehydrated with 95% and 70% methanol. The slides were subsequently blocked with 3% BSA at room temperature for 1 h. Following washing with 1 × PBS, scFvs were incubated with the cells at room temperature for 1 h. Finally, bound scFvs were detected using mouse anti-HA antibodies and visualized by donkey anti-mouse antibodies conjugated with FITC (Jackson ImmunoResearch Laboratories). Furthermore, nuclei were counterstained, staining with Hoechst solution (Sigma). The slides were examined with the Confocal Spectral Microscope Imaging System (TCS SP5; Leica).
Epitope definition
Five sequential peptides with overlapping regions based on the pre-S2 epitope (4) of HBsAg were designed for key residue definition. Each peptide was synthesized and conjugated with a biotin molecule at the C-terminal (Kelowna, Inc.). For binding analysis, a microtiter plate was precoated with recombinant streptavidin protein (1 μg/well) at 4°C overnight. After blocking with 3% BSA, the synthesized biotin-labeling peptides were added to the wells individually and incubated for 1 h at room temperature. After washing with PBST, scFvs were added to the wells and incubated for 1 h at room temperature. Finally, the scFv binders were detected, as described earlier.
Surface plasmon resonance
Peptide and antibody binding analysis was performed with the OpenSPR instrument (Nicoya Lifesciences, Kitchener, Canada). In total, 100 μL of the pre-S2 peptide (100 μg/mL) in PBS was provided as the target and immobilized on a streptavidin sensor chip. In operation, the running buffer was PBS, and a constant flow rate of 20 μL/min was used. ScFv S17 was dissolved in the running buffer at concentrations of 0.83–3.34 μM for the experiment. The sensor chip was regenerated using 10 mM glycine–HCl (pH 2) at a flow rate of 150 μL/min after each injection of scFv S17. Finally, the data were recorded and analyzed using the Trace Drawer software (Ridgeview Instruments, Uppsala, Sweden) as recommended by the manufacturer's protocol. The kinetic parameters of the interaction of scFv S17 and the pre-S2 peptide were investigated using software TraceDrawer according to a 1:1 binding model.
Molecular modeling
To further investigate the interaction of scFv S17 and the pre-S2 epitope, scFv S17 was modeled as the scFv scaffold, and the three-dimensional crystallographic structure of pre-S2 (NSTTFHQALLDPRVRGLYFPAGG, amino acids 123–145 of the pre-S region) was derived from the Protein Data Bank (PDB ID: 1WZ4). The homology model of scFv S17 was generated with the MODELER program in Discovery Studio v. 2017 (BIOVIA, Inc., San Diego, CA) by using the crystallographic structures of the anti-gp41 antibody gamma light chain (PDB ID: 3P30), the heavy chain of the antibody (PDB ID: 5AZE), and the antibody–antigen interface (PDB ID: 4FQJ) as structural templates. The complex structural model of the scFv S17-pre-S2 peptide interaction was developed by in silico antigen–antibody docking to obtain a S17-pre-S2 complex. A rigid-body docking program, ZDOCK (3), with default parameters, was used to search all possible binding modes for the antibody and antigen to obtain more accurate predictions for the two proteins by evaluating shape complementarity and desolvation energy. The leading 2,000 predictions from ZDOCK were fed into RDOCK, where they were minimized by CHARMm to improve the energy levels and eliminate clashes. Subsequently, the desolvation energy and electrostatics were recomputed by RDOCK. To further obtain a reliable complex structure in the solvated system, the best prediction from RDOCK was added with water molecules and optional counterions to allow the simulation of the solvent by minimizing and calculating the solvation model developed using Discovery Studio v 2017. The solvated system can be optionally minimized to eliminate the van Der Waals clashes. Finally, the rational antibody–antigen complex model satisfied the complementarity-determining regions (CDRs) of the scFv S17 antibody interacting with the active site of the pre-S2 antigen at the antigen–antibody interface.
Results
Generation and sequencing of the specific scFv
Human PBMCs were isolated from a person with history of HBV infection to generate a high-quality scFv library; the library complexity was 2 × 108. Four times of panning were performed to eliminate nonspecific binders and enrich specific clones. Furthermore, randomly selected scFvs were classified into two groups with the same sequence and are represented as scFv S17 and S27. The binding activities of both scFvs against HBsAg were analyzed using ELISA. As shown in Figure 1A, both scFvs showed markedly binding activity to HBsAg, but not to BSA. Moreover, their ability to recognize immobilized proteins was analyzed through Western blotting. The results revealed that scFvs S17 and S27 can clearly recognize recombinant HBsAg on the membrane (Fig. 1B). Furthermore, the VL and VH genes of the isolated scFvs were analyzed from ImMunoGeneTics website to determine the genetic diversity and variation of the CDRs. Sequencing results were aligned with the potential germline gene sequences of human immunoglobulins, as revealed in Table 1. The highest homology of scFv 17 to VL and VH germline genes was VL1–47 (68.1%) and VH1–8 (94.1%), whereas scFv S27 showed the highest homology to germline VL2–23 (87.9%) and VH3–30 (89.6%) (Table 1).
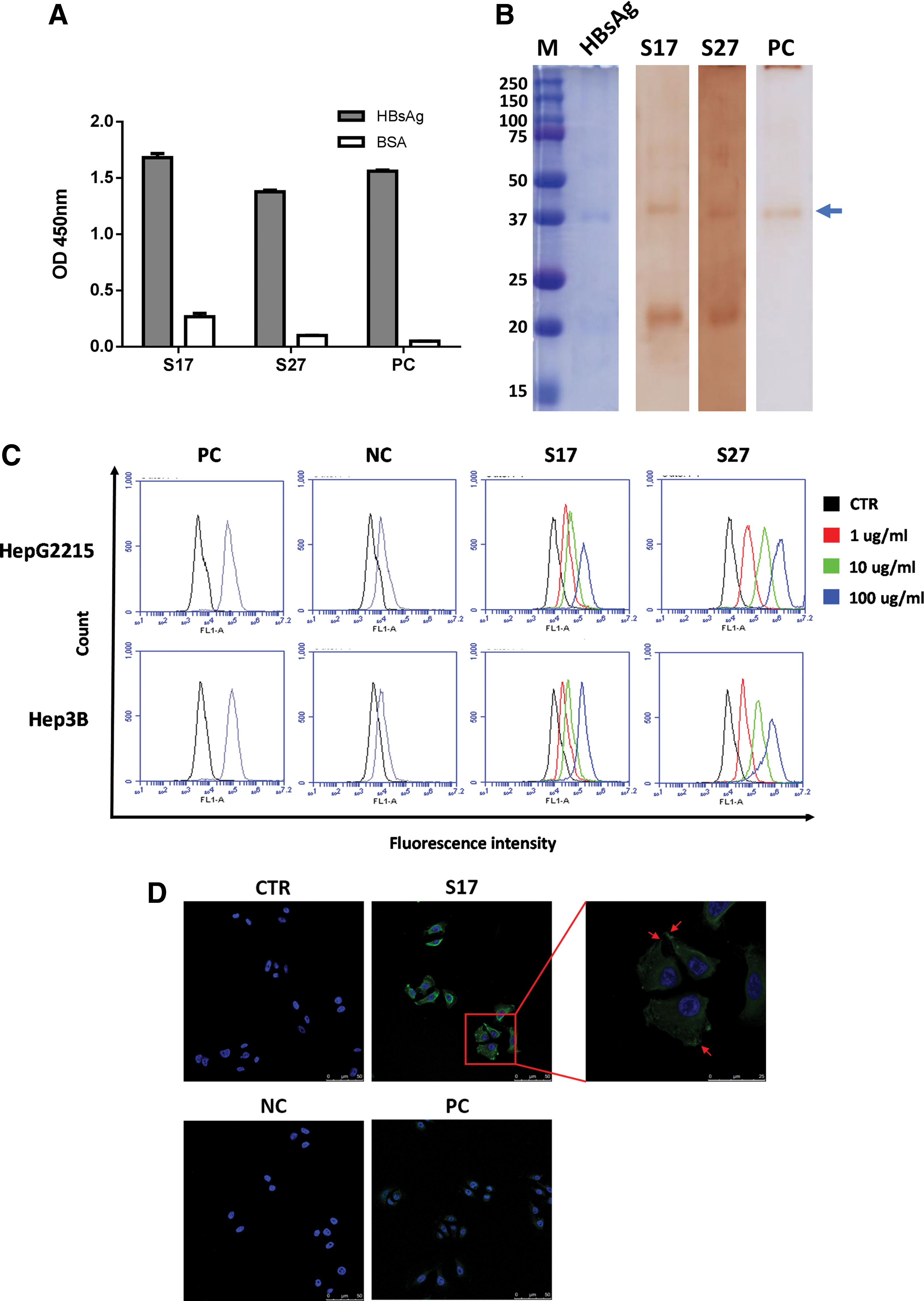
Characterization of anti-HBsAg scFv antibodies.
VH, heavy chain variable; VL, light chain variable.
Flow cytometry and immunocytochemical staining
Two HBV gene-incorporated cell lines, Hep2215 and Hep3B, were used to assess the binding reactivity of scFvs S17 and S27 against endogenous HBsAg expressed on the cell membrane through flow cytometry analysis (Fig. 1C). Significant binding signals were observed from both scFvs in a dose-dependent manner. To further confirm the binding of scFvs to HBsAg on the cell surface, scFvs were added to HepG2215 cells and subjected to immunocytochemical staining. Unexpectedly, scFv S27 was prone to precipitate at room temperature, thus showing high background in the staining (data not shown). The precipitation may be due to the instability of its scaffold. A commercial anti-HBsAg polyclonal antibody was used as the positive control to confirm the staining result, in which scFv S17 exhibited clear staining reactivity at the cell membrane (Fig. 1D). The binding signal of the cell extension position of HepG2215 cells revealed scFv S17 binding at the membrane position.
Epitope definition and binding affinity determination
To confirm the binding specificity of scFv S17 to the pre-S2 peptide, the pre-S1 epitope peptide (HQLDPAFGANSNNPD), which is included in the pre-S1 fragment, has been designed. As shown in Figure 2A, scFv S17 specifically bound to the pre-S2 peptide without crossbinding to the pre-S1 peptide. To confirm the key site of the pre-S2 epitope peptide on HBsAg recognized by the human scFv S17, five sequential peptides, including the pre-S2 peptide (amino acids 123–145; Fig. 2A) with overlapping regions, were synthesized for analysis. ELISA was performed to assess the binding reactivity of scFv S17 to these peptides, for which the pre-S2 peptide showed the highest binding reactivity (Fig. 2B). ScFv S17 can bind to peptide 123–145 and peptide 134–155. Minimal epitope recognized by scFv S17 should be located in the overlapping region of amino acids 134–145. The result is similar to that of scFv S27 (Supplementary Fig. S1; Supplementary Data are available online at
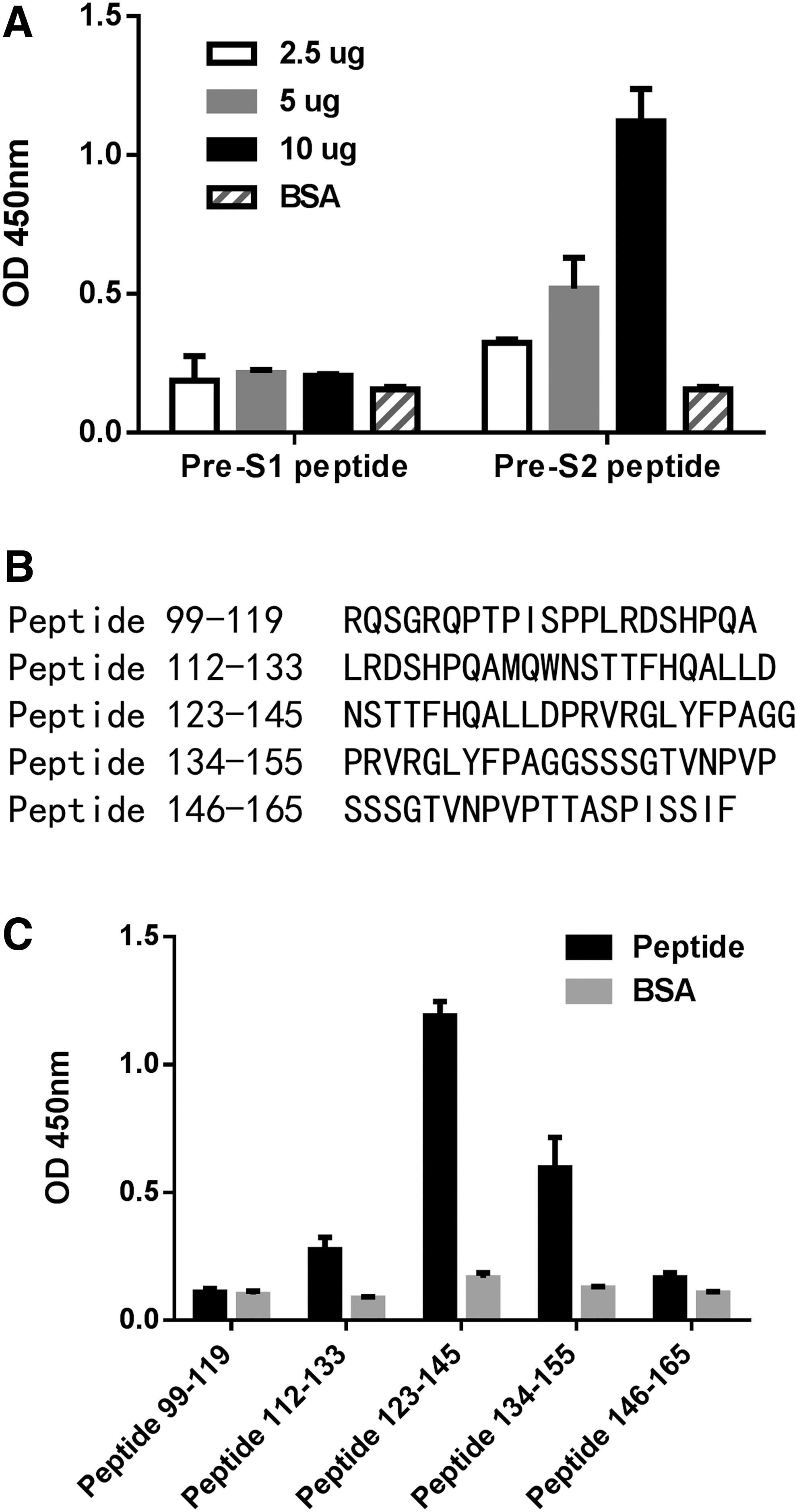
Epitope mapping of human scFv S17.
KD , dissociation constant; scFv, single-chain variable fragment.
Interaction of scFv and the pre-S2 epitope
The homology model of scFv S17 was generated by using structural templates with multiple sequence alignment of variable domains of the VL and VH regions of scFv S17 with 3P30, 5AZE, and 4FQJ antibodies (Fig. 3). The antibody–peptide docking predicted that the CDRs of scFv S17, namely L1, L3, H2, and H3 (but not L2 and H1 loops), were involved in the interaction of the pre-S2 epitope and scFv S17 (Fig. 4A). The pre-S2 peptide has 23 residues (amino acids 123–145 of the pre-S region), as inferred from Lue10 to Ala21 being key residues of the scFv S17 epitope. Ten residues of the CDR loops of scFv S17 were in contact with the pre-S2 epitope. The mechanism of epitope recognition involves five hydrogen bonds, five hydrophobic interactions, and two ion interactions.
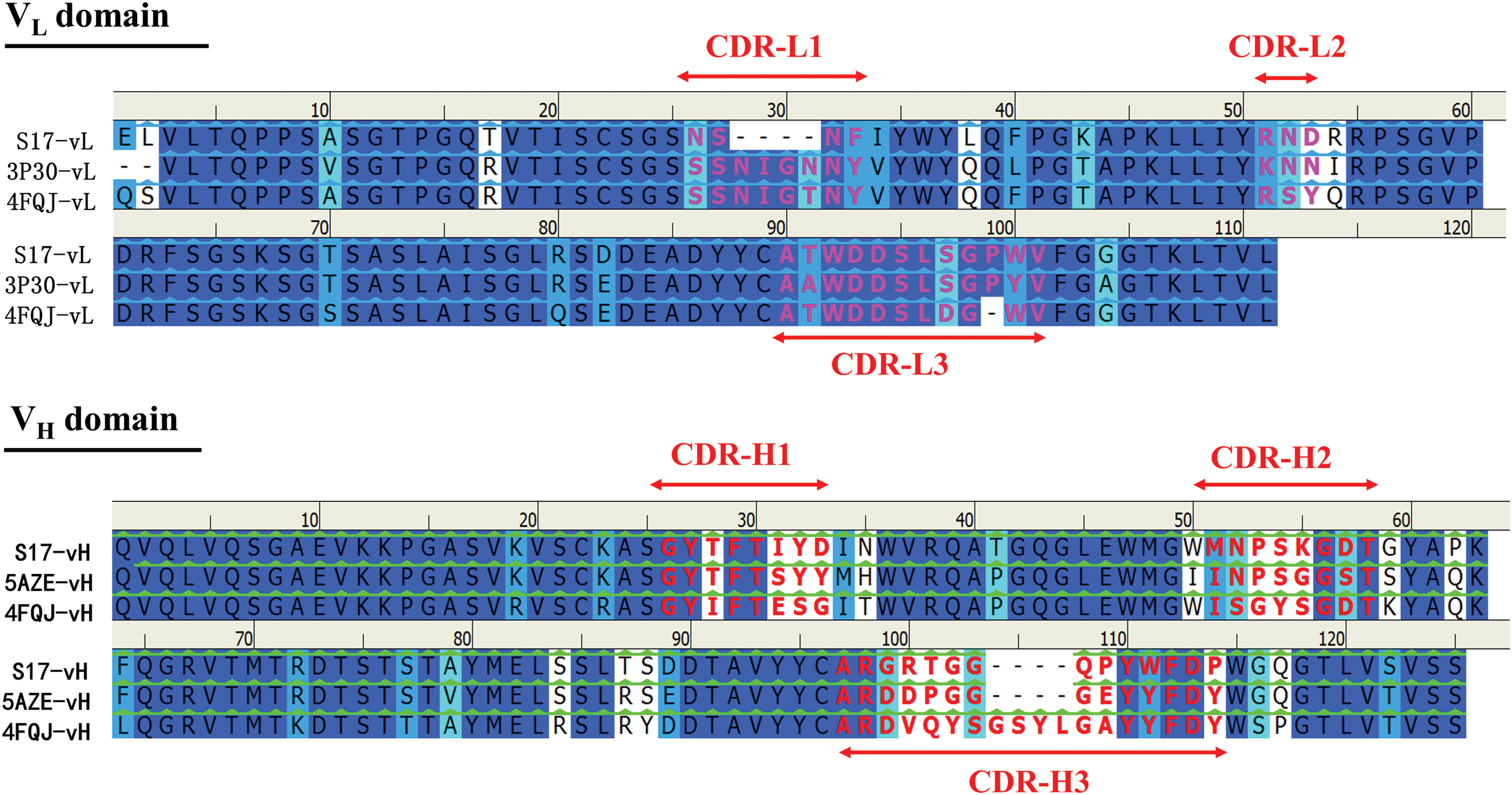
Multiple sequence alignment of variable domains of the VL and VH regions of the scFv S17 with 3P30, 5AZE, and 4FQJ antibodies. The background of the residues is colored according to sequence similarity. Deep blue indicates conserved residues in all sequences. The color scheme from deep blue to light blue and cyan corresponds with high and low conserved residues, respectively. The background of the residues, indicated in white, represents no similarity. The residues of the CDRs are indicated in bold in red. CDRs, complementarity determining regions; VH, heavy chain variable; VL, light chain variable. Color images available online at
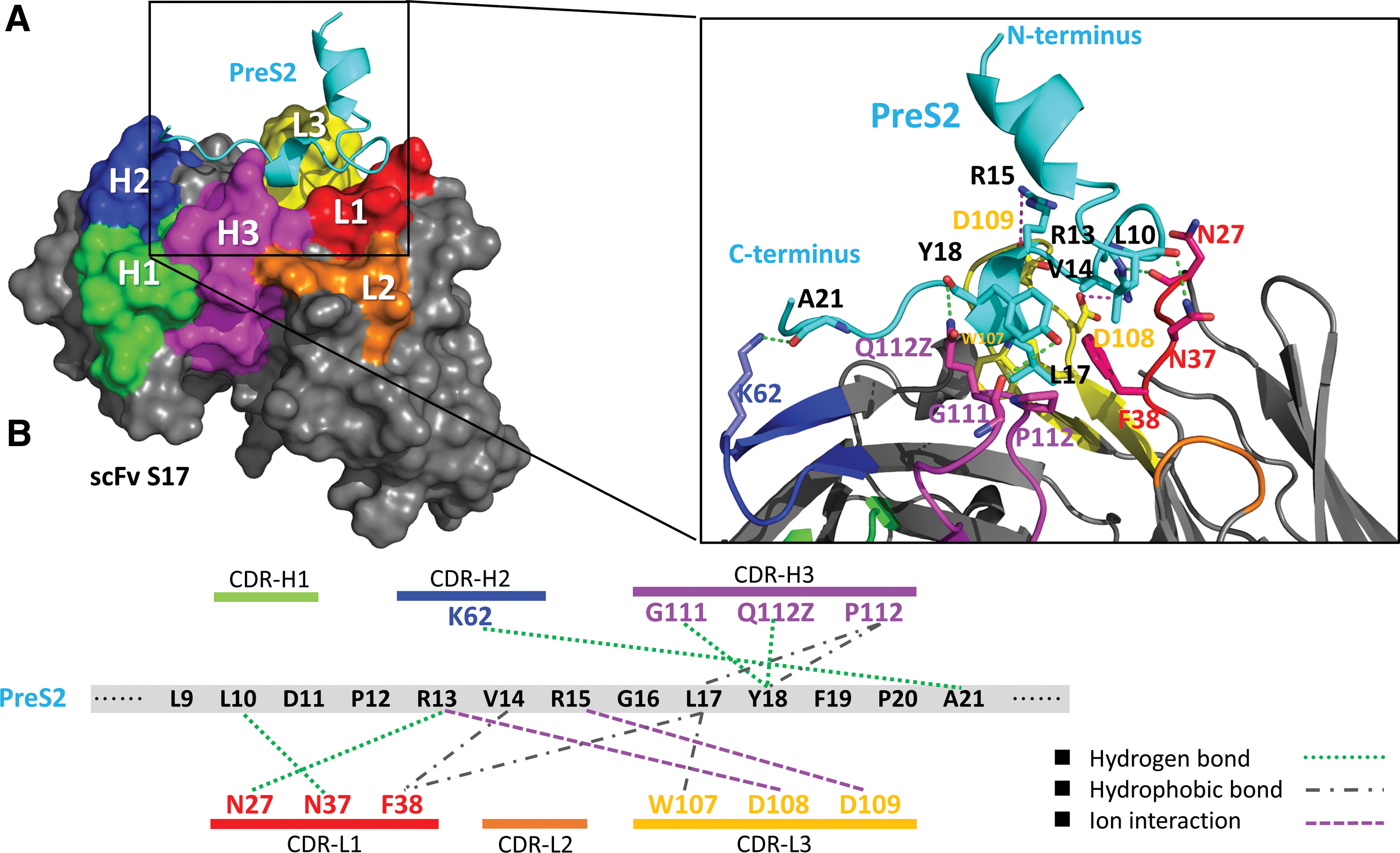
Interactions of scfv S17 with pre-S2 epitope.
By putative analysis, the only interaction on the CDR–H2 loop was a hydrogen bond between the side chain of Lys62 and the main chain of Ala21. The CDR–H3 loop interacted with the pre-S2 epitope on two residues, Leu17 and Tyr18. The side chain of Pro112 acts on the side chains of Leu17 and Tyr18 through hydrophobic interactions. Meanwhile, the main chain of Gly111 and the side chain of Gln112Z interact with the side and main chains of Tyr18, respectively, through hydrogen bonding. Three interactions were generated by the light chain of scFv S17 and the pre-S2 epitope, namely, hydrophobic interactions, hydrogen bonds, and ion interactions. The side chains of Asn27 and Asn37 on the L1 loop interact with the side chain of Arg13 and the main chain of Leu10, respectively, through hydrogen bonding. Moreover, the side chain of Phe38 interacts with the side chains of Val14 and Leu17 through hydrophobic interactions. Similarly, on the CDR–L3 loop, hydrophobic interactions occurred between the side chains of Trp107 and Leu17. The side chains of Asp108 and Asp109 on the CDR–L3 loop interact with those of Arg13 and Arg15, respectively, through ion interactions (Fig. 4B).
Moreover, the interactions between scFv S27 and the pre-S2 epitope have also been predicted and analyzed by molecular modeling. The homology model of scFv S27 was generated by structural templates with multiple sequence alignment of variable domains of the VL and VH regions of the scFv S27 with 5U15, 5U68, and 3H42 antibodies (Supplementary Fig. S2). Similarly, scFv S27-pre-S2 docking revealed that the CDRs of scFv S27, namely L1, L3, H1, H2, and H3 (but not L2 loop), were involved in the interaction of the pre-S2 epitope and scFv S27 (Supplementary Fig. S3A). Meanwhile, their interactions are shown in Supplementary Figure S3B.
Discussion
In this study, we described the isolation of anti-pre-S2 scFvs from a person with HBV infection history. It is noteworthy that high anti-pre-S2 antibody titer was found in the serum of the donor, implying the importance of the pre-S2 epitope for neutralizing antibody induction against viral infection. Therefore, the purpose of this study is to isolate the specific antibodies directly against the pre-S2 epitope by phage display technology and characterize these anti-pre-S2 antibodies produced by somatic mutation. Generally, neutralizing antibodies produced by vaccination is the optimal strategy to prevent HBV infection. Because anti-pre-S2 antibody produced by the humoral immune system is proven to be efficient against HBV infection (6), the pre-S1 fragment and pre-S2 fragments are included in the HBV vaccine to enhance the immune response (13,17). These neutralizing antibodies can recognize critical epitopes on the surface antigen that are required for virus infection. The binding affinity of the isolated scFv S17 to the pre-S2 peptide was determined to be 4.2 × 10−8 M by the SPR system (Supplementary Fig. S4). The flexible scaffold of scFv may affect and reduce the affinity, and therefore result in scFv S17 having a moderate binding affinity. The actual complete IgG molecular structure in the donor should have higher affinity. Moreover, phage-derived scFv may represent the artifacts of the recombination of the VH and VL regions during library construction; this may be another reason for the issue. Despite these uncertain factors, the isolation of the specific antibodies by phage display system and the studying on their function are still widely accepted. We believe that characterization of the isolated scFv against the neutralizing epitopes can still provide useful insights into the interaction mechanism.
In the panning process, adequate increase in the output (eluted) phage number compared with the corresponding number in the original library implies successful enrichment (2). Due to the affinity selection-based panning, lower enrichment levels may be insufficient to eliminate the nonspecific and low-affinity binders, and may thus result in the outgrowth of unwanted phages after amplification. In this study, the eluted output phage number improved by approximately 30-fold after panning, which is sufficient to enrich specific binders (Supplementary Fig. S5A). Moreover, increasing signals from the phages, as detected using ELISA, showed that the ratio of enriched phage binders increased in the amplified library (Supplementary Fig. S5B). Notably, the output phage number showed an instant increment after the third round of panning, although it had slightly decreased in the second round. This result is in accordance with our previous study and other researches (7,12,15).
Antibodies that were isolated in this study are targeting to pre-S2 epitope directly. The epitope (residues 123–145) in the pre-S2 region of HBV has been published and known to be associated with antibody neutralization (4). It is reasonable to speculate that the isolated scFvs have neutralization potential against HBV infection. In this study, the peptide docking analysis of the antibody–antigen interaction indicated that the secondary structure of 23 specific amino acids from the pre-S2 epitope formed a hook-like structure because the epitope had two proline molecules (Pro12 and Pro20, which formed the minimal epitope), including amino acids 134–142 of the pre-S region (PRVRGLYFP) for anti-pre-S2 scFv S17. The peptide-binding sites (CDR–L1,–L3,–H2, and –H3) of scFv S17 presented a bowl-like shape. ScFv S17 interacted with pre-S2 through three binding interactions: hydrogen bonds, hydrophobic interactions, and ion interactions. We observed that two positively charged arginine residues in the PRVRGLYFP sequence (amino acids 134–142 of the pre-S region) dominated the ion interaction. Two arginine molecules of the RVR sequence of pre-S2 (amino acids 135–137 of the pre-S region) correspond to two negatively charged aspartate residues on the CDR–L3 loop of scFv S17 (Asp108 and AspD109). The docking model showed the two ion interactions as the major driving forces for scFv S17 and pre-S2. However, one previous study (11) did not report that the specific RXR sequence of pre-S2 dominates the antibody–antigen interaction. Moreover, we observed hydrogen bonds at the binding site around the antibody–peptide interface. Hydrophobic interactions in the unexposed binding site between scFv S17 and pre-S2 might further stabilize the antibody–peptide complex. Compared to the interactions of scFv S27 and scFv S17 with pre-S2, CDR-L1 and CDR-H3 of two scFvs were found to interact with pre-S2 through hydrophobic interactions and hydrogen bonding. Especially, scFv S17 has two putative ionic interactions, more than scFv S27 in CDR-L3. In conclusion, this study characterizes the specific anti-pre-S2 human scFv antibodies, and the molecular modeling structural investigation is in line with the experiments.
Footnotes
Acknowledgments
We thank Prof. Shiow-Lin Pan (The Ph.D. Program for Cancer Molecular Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan) with providing human cDNA for antibody library construction. This study was supported by a start-up fund from Taipei Medical University (TMU103-AE1-B19) and the Ministry of Science and Technology in Taiwan (MOST 104-2320-B-038-012 and MOST 105-2628-B-038-007-MY3).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.