
Abstract
Epitope escape from HIV-1-targeted CD8+ cytotoxic T lymphocyte (CTL) responses occurs rapidly after acute infection and contributes to the eventual failure of effective immune control of HIV-1 infection. Because the early CTL response is key in determining HIV-1 disease outcome, studying the process of epitope escape is crucial for understanding what leads to failure of immune control in acute HIV-1 infection and will provide important implications for HIV-1 vaccine design. HIV-1-specific CD8+ T lymphocyte responses against viral epitopes were mapped in six acutely infected individuals, and the magnitudes of these responses were measured longitudinally during acute infection. The evolution of autologous circulating viral epitopes was determined in four of these subjects. In-depth testing of CD8+ T lymphocyte responses against index and all autologous-detected variant epitopes was performed in one subject. Among the four individuals examined, 10 of a total of 35 CD8+ T cell responses within Gag, Pol, and Nef showed evidence of epitope escape. CTL responses with viral epitope variant evolution were shown in one subject, and this evolution occurred with and without measurable CTL responses against epitope variants. These results demonstrate a dynamic period of viral epitope evolution in early HIV-1 infection due to CD8+ CTL response pressure.
Introduction
CD8+
However, CTL-mediated immunity does not typically lead to containment or eradication of HIV-1 infection. The reason for this failure of CTL-mediated immunity is multifactorial (34,37,10) and involves the evasion of CTL responses by viral epitope escape (30). Understanding what factors may decrease the likelihood of viral escape, particularly in early HIV-1 infection, would help explain why the CTL immune response fails to lead to successful immunity and would also help advance the development of a successful vaccine (36).
Viral epitope escape of the CTL response occurs as a result of the high mutation rate of the HIV-1 genome during viral replication, estimated to be equivalent to every single- or double-point mutation of the HIV-1 genome sequence to be produced every day in the viral pool of an infected individual (9). Although mutations may lead to significant effects on viral protein structure and function, resulting in a fitness cost to certain variant species (24), sufficient variations in viral sequences occur to produce epitope escape of CTL responses (26). Epitope escape has been shown to occur readily in early HIV-1 infection models (3), and as early as 16 days after the onset of symptoms of primary HIV-1 infection (7) in humans.
Escape by reduced recognition of the T cell receptor (TCR) is limited by CTL promiscuity, or the ability of CTL responses to recognize and target variant epitopes. Given the stochastic development of the TCR, each CTL clone has distinct promiscuity, such that each clone can target a unique set of epitope variants (42). A CTL response against a particular epitope can also involve more than one T cell clone, and the number of clones comprising a response shared between individuals can vary (5,40). The sum total of all CTL clones specific to a given epitope represents the clonal breadth of a CTL response to a particular epitope. Because the number of variant epitopes targeted would be greater, CTL responses with larger breadth may better restrict viral escape. Escape may also be contained by the formation of new CTL responses with distinct clonal repertoires developing against variant epitopes selected under CTL immune pressure (4).
Studying the viral epitope evolution in early infection and investigating the effect of CTL clonal breath and new CTL response development against these epitopes will add to the understanding of what constitutes a more robust CTL response, aiding in HIV-1 vaccine design and development. In this exploratory study, we systematically identified CD8+ T lymphocyte responses during acute infection and correlated the progression of these immune responses to the evolution of viral epitope variants. We sought to expand on prior studies by tracking the magnitude of CTL responses against targeted epitopes as well as detected autologous variant epitopes to explore how clonal breadth may influence the development of epitope escape and give further insights into the dynamics of this process. Our results show a detailed picture of the CD8+ CTL response during a period of active viral evolution in acute HIV-1 infection.
Materials and Methods
Study subjects
Six acutely infected individuals were studied, each with detectable level of HIV-1 virus in blood and negative detuned ELISA and/or Western blot at the time of enrollment. Peripheral blood mononuclear cells (PBMCs) and plasma were collected at multiple time points from each subject beginning from the time of enrollment up to 13 weeks afterward. HLA serologic equivalent testing of subjects was performed with sequence-specific oligonucleotide hybridization.
Cell culture
All cell culture utilized medium consisting of RPMI 1640 (Sigma) supplemented with 10% heat-inactivated fetal calf serum, 10 mM N-2-hydroxyethylpiperazine-N9-2-110 ethanesulfonic acid, and 2 mM glutamine, unless otherwise noted.
Nonspecific expansion of CD8+ T lymphocytes from PBMC
CD8+ T lymphocytes were nonspecifically expanded from PBMC in culture containing 50 U/mL of recombinant human interleukin-2 (NIH AIDS Reference and Reagent Repository) and CD3:CD4 bispecific antibody (39) for 2 weeks, consistently resulting in CD3+ lymphocyte populations that were >90% CD8+.
Detection of CTL responses with ELISpot
Ninety-six-well nitrocellulose filter plates (Millipore, Burlington, MA) were coated with monoclonal anti-IFN-gamma antibody (Pharmingen; BD Biosciences, San Diego, CA) and plated with up to 2 × 105 expanded CD8+ T lymphocytes per well in medium. Cells were treated with a library of consecutive 15-mer HIV-1 peptides overlapping by 11 amino acids and spanning all HIV-1 Clade B consensus proteins (NIH AIDS Research and Reference Reagent Repository) or specific minimal peptide epitopes. Minimal peptide epitopes were selected based on previously known HLA class I antigens or by predicting the most likely minimal epitope within a positively screened 15-mer peptide using the online programs SYFPEITHI Epitope prediction (31) and Bioinformatics and Molecular Analysis Section (Bimas) (29). Cells were treated with peptides at a concentration of 5 μg/mL. Each plate also contained four negative control wells without peptide stimulation, and a positive control well treated with 1 mg/ml phytohemagglutinin (PHA). After incubation for 12–16 h, cells were stained with a biotinylated second IFN-gamma-specific antibody labeled with streptavidin peroxidase. Each well was then treated with streptavidin peroxidase and developed with a peroxidase color reagent. Spots that formed on well filters representing individual IFN-gamma-secreting CD8+ T lymphocytes were counted as spot-forming units (SFU) with an automated counter (Cellular Technologies Limited, Cleveland, OH) and confirmed manually. Positive responses were defined as having at least 60 SFU per million cells and more than the mean plus three times the standard deviation of the negative wells (14). Peptides were screened with pools of 12–16, and individual CTL responses were identified with individual peptides.
Detection of autologous viral variants within Gag-Pol and Nef sequences
Creation of an NL4-3 ΔGag-Pol cloning vector
The p83-2 plasmid containing the 5′ genomic portion of NL4-3 (1) served as the starting template for the construction of a target vector for rapid cloning of primary Gag-Pol sequences into the NL4-3 backbone. First, site-directed mutagenesis (QuikChangeIIXL; Agilent, Westlake Village, CA) was performed to change nucleotides 781–786 into an StuI site (AGG^CCT). Second, nucleotides 5,044–5,049 were mutagenized to contain an HpaI site (GTT^AAC), which also created a premature stop codon to eliminate virus production from plasmid without the desired Gag-Pol replacement. This formed the p83-2ΔGag-Pol vector. In addition, a whole genome version was constructed by creating an NruI site at the junction of the HIV-1 insert and the plasmid backbone of p83-2ΔGag-Pol. After cutting with NruI and EcoRI (New England Biolabs, Ipswich, MA), the resulting HIV-1 fragment was ligated into either p83-10/HSA or p83-10/HSA-HA plasmids containing the 3′ end of the HIV-1 genome with reporter genes in the Nef locus as previously described (2). This formed the pNL4-3/HSAΔGag-Pol and pNL4-3/HSA-HAΔGag-Pol whole genome plasmids. All modifications were confirmed by sequencing.
HIV-1 Gag-Pol amplification and cloning into NL4-3
Viral RNA was isolated from plasma using the QIAamp UltraSens viral isolation kit (Qiagen, Germany). Viral RNA was then reverse transcribed and the Gag-Pol region of the HIV-1 genome was amplified using multiple primers spanning these regions. Hemi-nested PCR was performed using KOD hifidelity DNA polymerase (EMD, Billerica, MA). The first PCR used primers 737F and AGPolRnst, and the second PCR used primers F2nst and AGPolRnst (Supplementary Table S1; Supplementary Data are available online at
Full-length Gag-Pol sequencing and analysis
Nine or more full-length Gag-Pol clones per time point were isolated per subject by picking bacterial colonies after transformation of the final plasmid constructs. Full-length Gag-Pol DNA sequencing was completed with 10 primers (Supplementary Table S1) using the Big Dye 3.1 sequence terminator kit (Applied Biosystems, Carlsbad, CA) on an AB3130 genetic analyzer and edited with ChromasLite. Edited sequences from each clone were assembled into the full-length Gag-Pol sequence, translated, and aligned to the Los Alamos HIV Sequence Database 2010 clade B consensus sequence, manually adjusted using BioEdit, and analyzed for fixed amino acid changes.
The protocol for Nef sequences is described in De La Cruz et al. (11). Detection of autologous viral sequences was not performed on subjects 3 and 6 due to unavailability of samples.
The materials, methods, and study design were reviewed and approved by the University of California–Los Angeles Institutional Review Boards (IRBs) to ensure protection of human subjects.
Results
Each of the six subjects demonstrated detectable viral blood levels and had negative detuned ELISA and/or Western blot tests at the day of enrollment (day 0). The HLA profiles of subjects are shown in Supplementary Table S2. PBMC and plasma samples were obtained spanning a time frame of up to 13 weeks. The plasma viral load and CD4 T cell counts during the testing time period for each subject are shown in Figure 1.
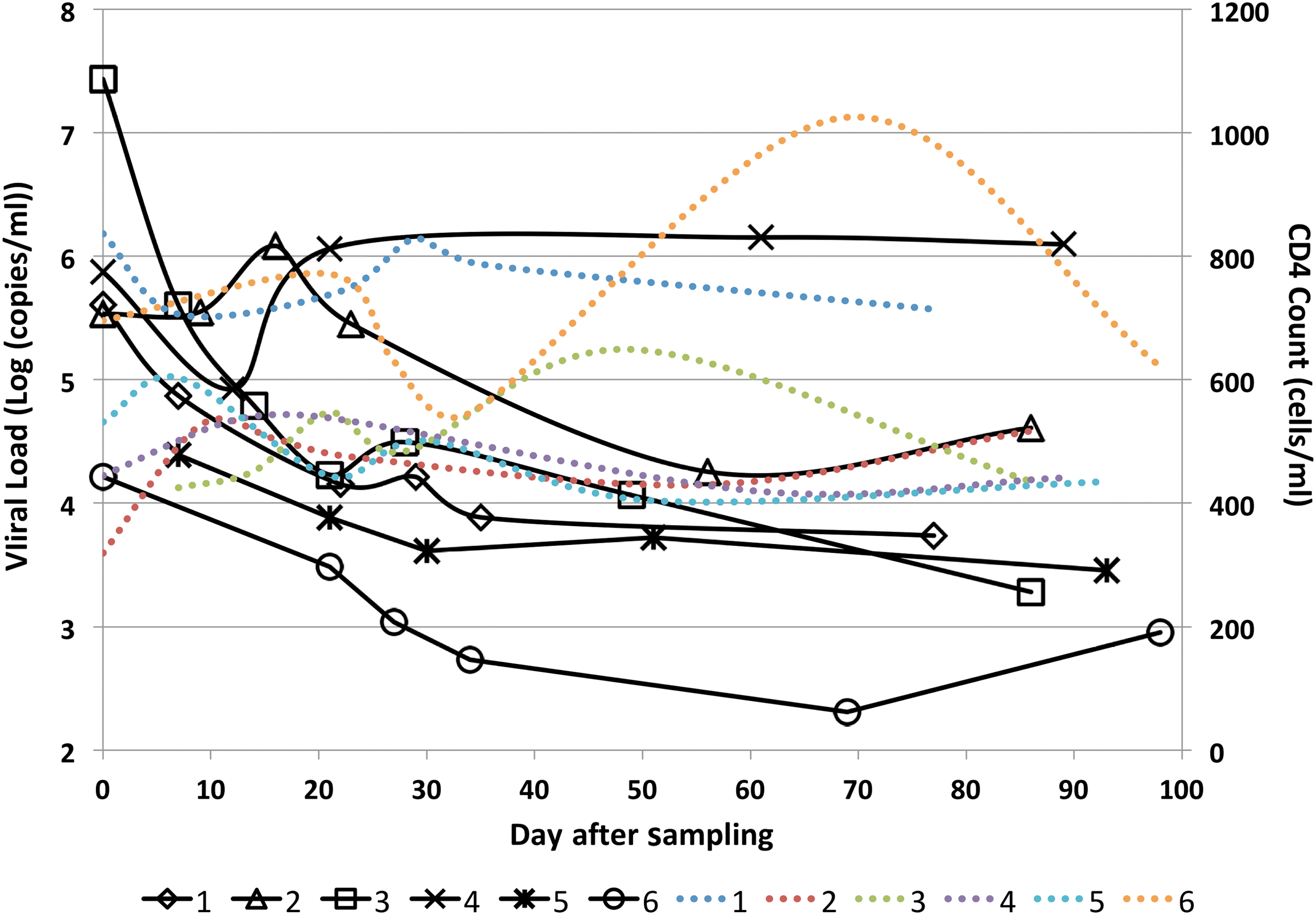
Viral blood levels and CD4 T cell counts of the six subjects measured at several time points over the course of the sampling period. Black lines represent viral load (in Log [copies/mL]) of different subjects, and dotted color lines represent CD4 T cell counts (in cells/mL) of different subjects; lines corresponding to number of subjects are shown in the legend.
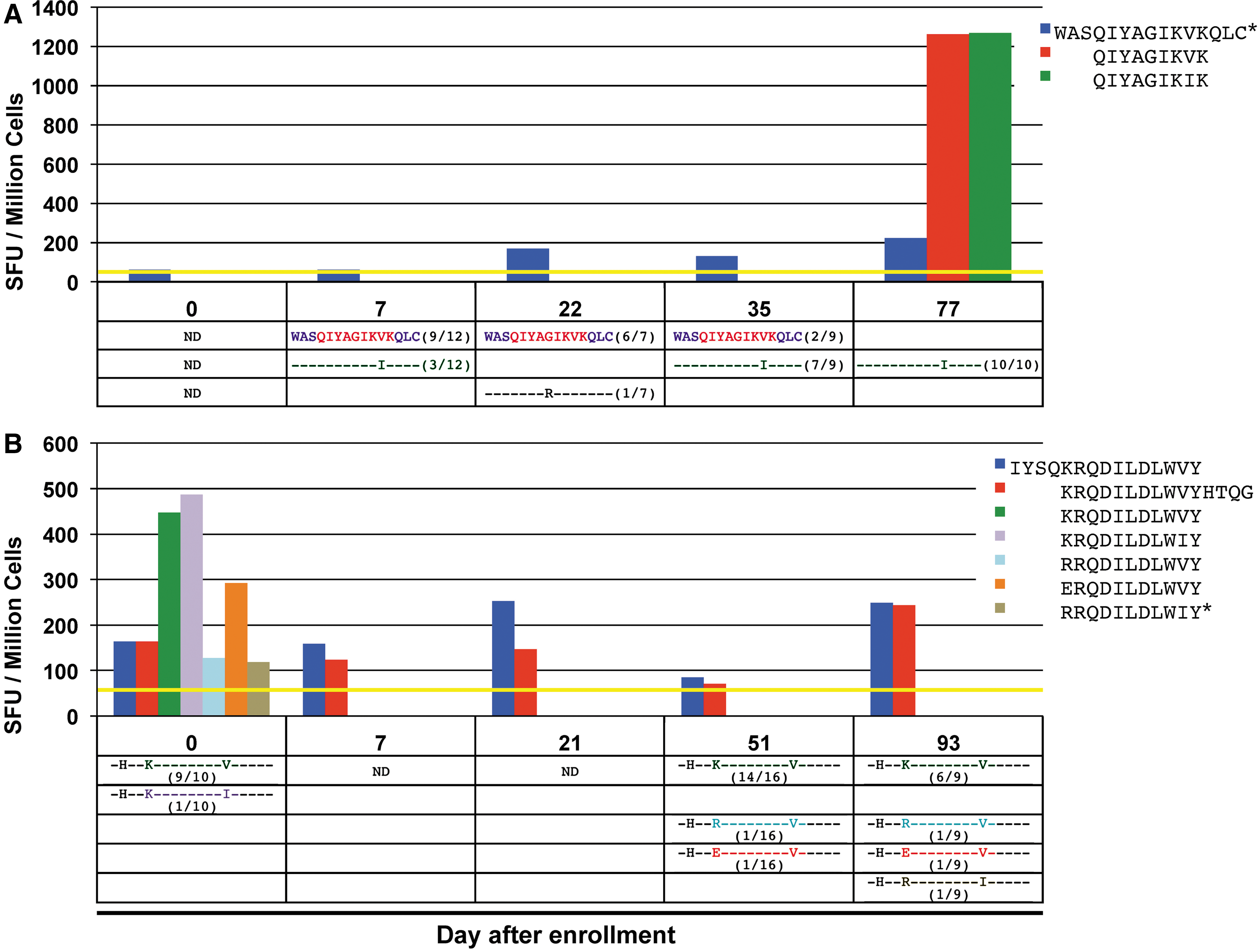
ELISpot mapping in these subjects revealed 54 distinct responses against screening 15-mers (after accounting for overlaps in amino acid sequences) and 32 distinct responses against previously known epitope antigens. Fourteen of the previously known epitope antigens were located within screening 15-mer peptide sequences, representing the corresponding targeted minimal epitope. Five more minimal epitope antigens were identified within Gag, Pol, and Nef by testing the SYFPEITHI or Bimas-predicted minimal epitopes within the remaining positively screened Gag, Pol, and Nef 15-mer peptides (marked in red in Supplementary Fig. S1). Figure 2A (top) shows a representative CTL response against a peptide (WASQIYAGIKVKQLC) in subject 1 measured with five sampling points over a span of 77 days.
The location of each final determined peptide antigen for all six subjects is shown in Figure 3, with the number of responses within each HIV-1 protein given in Table 1. Each epitope region was defined as a positively screened 15-mer peptide, an 8–11 amino acid sequence overlap between two positively screened 15-mer peptides, or a determined minimal epitope. The number of responses varied between each individual from 4 to 26, and the majority were directed at targets within Gag-Pol, Nef, and Env.
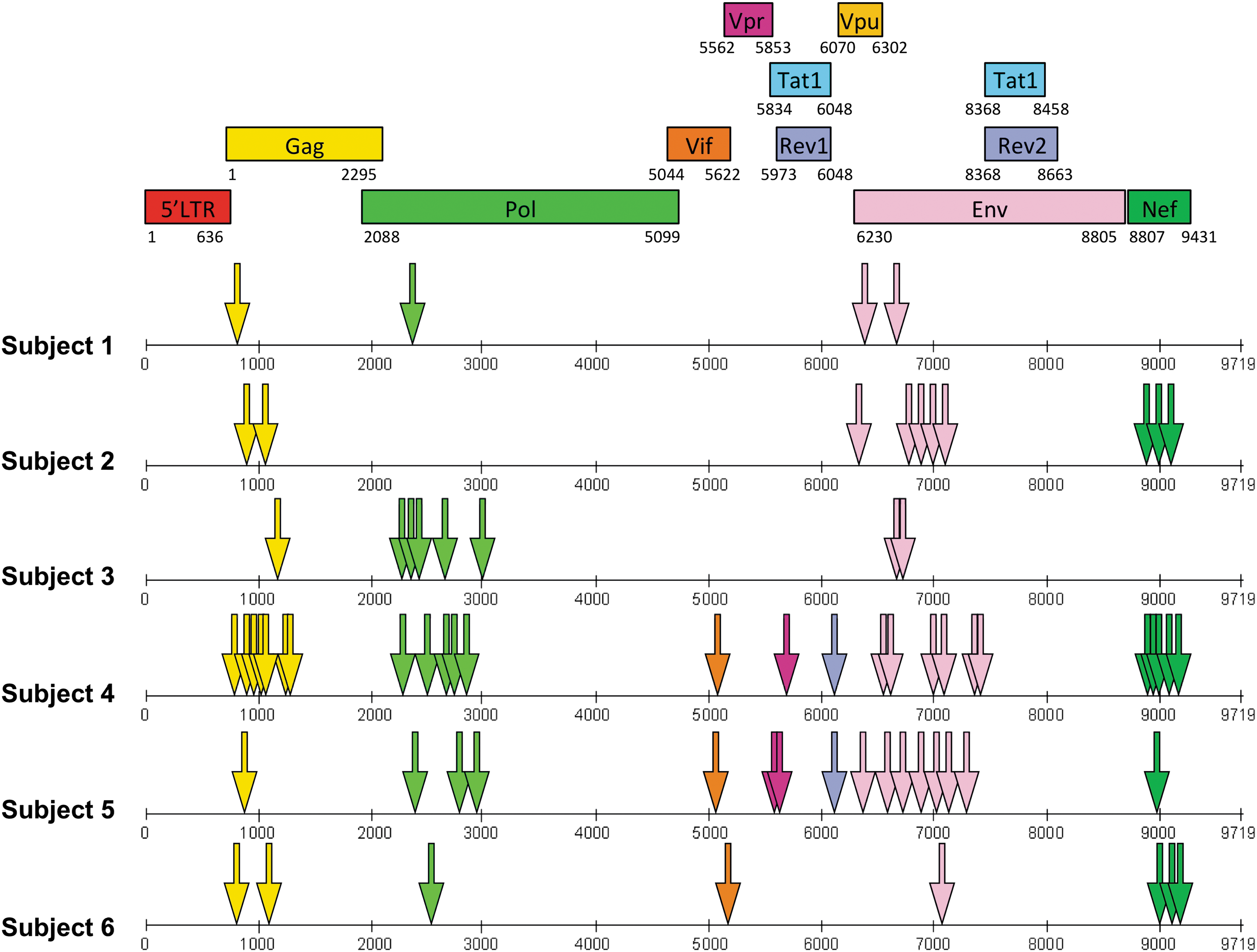
Location of each targeted peptide antigen (arrows) for all six subjects in the nucleotide sequence of HIV-1 (x-axis); colors correspond to the specific protein sequence map describing which protein each target peptide is located.
ND, not done.
For each PBMC sample in subjects 1, 2, 4, and 5, the corresponding plasma was isolated and autologous viral epitope variants within Gag, Pol and Nef-targeted epitopes were sequenced. Sequences were obtained at time points spanning at least 70 days (Supplementary Fig. S1). The frequencies of each detected variant were determined as shown for the representative CTL response against WASQIYAGIKVKQLC in Figure 2A (bottom).
In subjects 1, 2, 4, and 5, 10 of the 29 total responses within Gag, Pol, and Nef showed a decrease in the frequency of the most abundant initial epitope (index epitopes) over time (Table 1 and Supplementary Fig. S1), indicating that CTL pressure could be leading to epitope escape (instances where only one variant epitope sequence was noted at the last time point were excluded from this count). For example, in subject 1, there was a clear evolution of the index epitope QIYAGIKVK to the variant QIYAGIKIK, which becomes the only epitope detected at the final time point (Fig. 2A bottom). Equivalent CTL responses to both index and variant epitopes were detectable at the final time point. In subject 5, there was a decrease in frequency of the index epitope KRQDILDLWVY and the appearance of multiple different variants at the last two time points (Fig. 2B bottom). Interestingly, there were detectable CTL responses against these later variants on day 0 along with relatively strong responses against the index epitope and variant KRQDILDLWIY (Fig. 2B top).
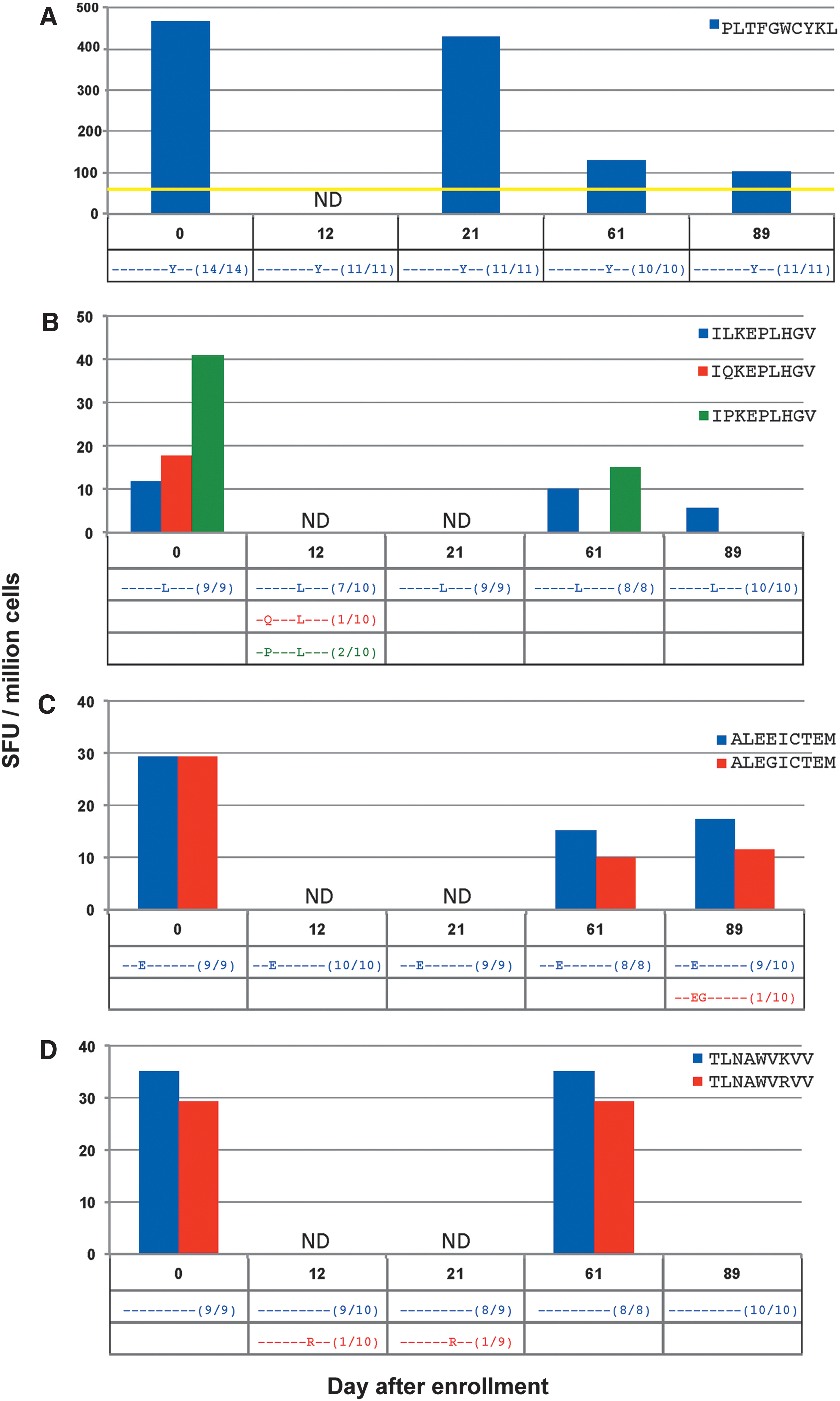
To create a comprehensive examination into epitope evolution and its relationship to CTL pressure against the index and all variant epitopes, ELISpot testing was repeated on all detected minimal viral epitope sequences in subject 4 (sample availability limited this analysis to only this subject). Twelve responses were tested that revealed several different scenarios (Fig. 4).
(
Responses where no variants were detected
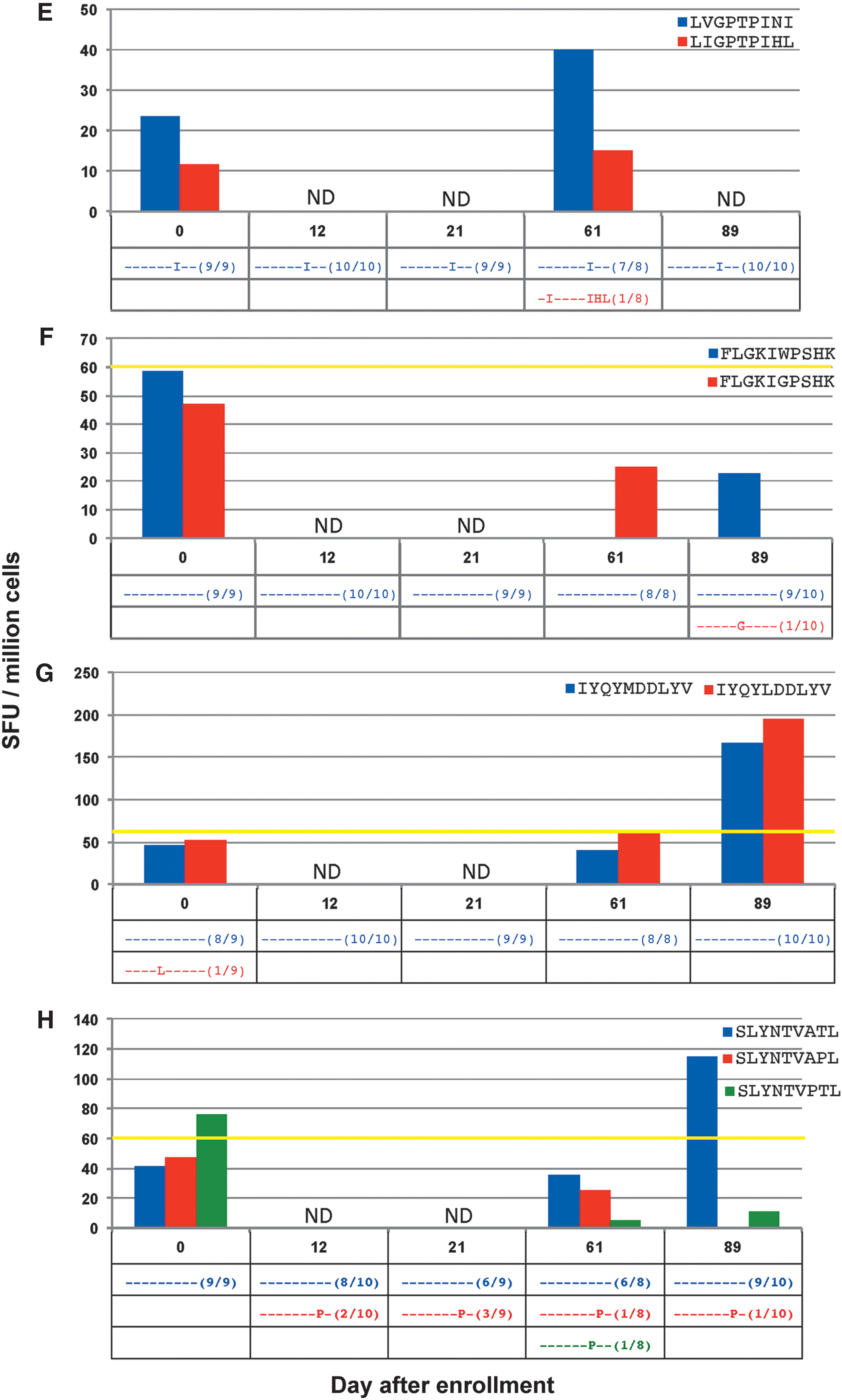
In the response against minimal epitope PLTFGWCYKL (Fig. 4A), no epitope variants were detected at any time point. Although the magnitude of the CTL response decreased, it was still detectable against the epitope until the last time point measured.
Instances where detection of the initial positive screening response was not replicated upon testing of variants
In CTL responses against the minimal epitopes ILKEPVHGV (Fig. 4B), ALVEICTEM (Fig. 4C), TLNAWVKVV (Fig. 4D), LVGPTPVNI (Fig. 4E), and FLGKIWPSHK (Fig. 4F), no measurable CTL responses against the initially screened index epitopes were detected during testing of epitope variants. In four of these responses, any variant epitopes found were present in low frequencies (only one out of all cloned sequences), and with the exception of the response against TLNAWVKVV, no variant was detected at more than one time point.
Instances where index CTL response was replicated, but no clear pattern of epitope variants in relation to CTL responses noted
In the response against the minimal epitope IYQYMDDLYV (Fig. 4G), one variant was seen on day 0 at low frequency. A response against the variant, IYQY
Instances with persistent CTL responses against index epitopes but no responses seen against epitope variants
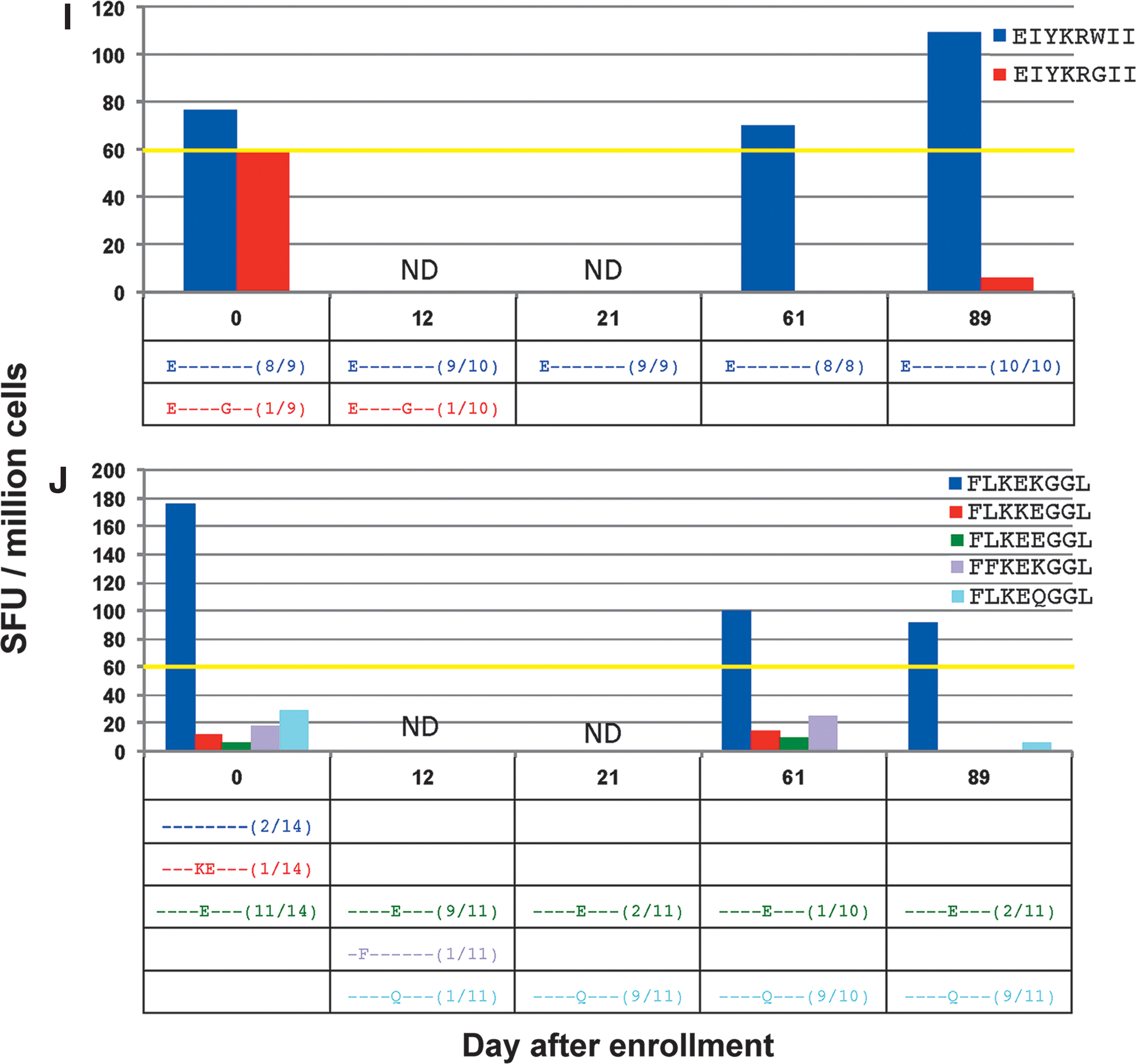
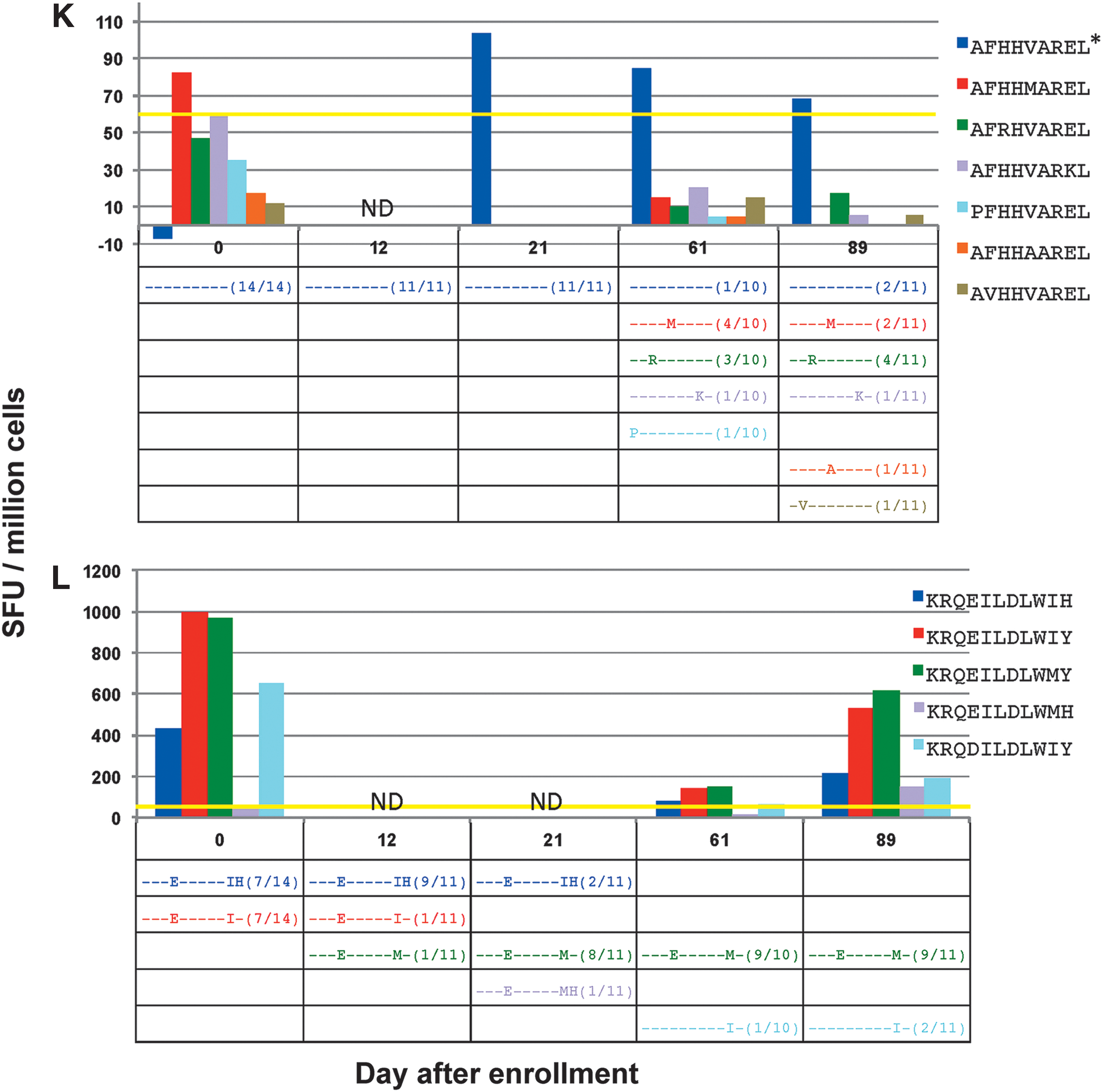
In CTL responses against the minimal epitopes EIYKRWII (Fig. 4I), FLKEKGGL (Fig. 4J), and AFHHVAREL (Fig. 4K), responses against the index epitopes could be confirmed, but no consistent CTL responses against variant epitopes were seen. In the case of EIYKRWII, the detected variant, EIYKR
Instances where CTL responses were seen against variants that persisted
One CTL response was noted against two variants at equal frequency on day 0, KRQEILDLWIY and KRQEILDLWIH (Fig. 4L). These variants were no longer detected by day 61, and by day 89, only two other variants remained, KRQEILDLW
Discussion
The level of virus decreased and stabilized in most subjects, indicating that their clinical time frame reflects Fiebig stage IV or V. The fall and then rebound of viral load seen in subject 4 is not typical of early infection.
Consistent with prior findings (7,13,23), evidence of epitope escape in acute infection was seen in multiple CTL responses, and the number of targeted epitopes varied between individuals (12,16,22). Similar to results from a study by Liu et al. (22) that examined viral CTL-targeted epitope dynamics in acute infection, a greater proportion of escape was seen in Nef-targeted epitopes within the 100 days following Fiebig stage I/II. However, our results differ in that a larger proportion of targeted epitopes were seen in Pol (21% of the total in this study vs. 3% in Liu et al.), and a smaller rate of escape occurred for epitopes within Gag, Pol, and Nef proteins within this time period (34% in this study vs. 53% in Liu et al.) despite a stricter definition of escape being utilized (<50% of transmitted/founder viral sequence detected). These inconsistencies are certain to be influenced by differences in methodologies, as the Single Genome Amplification utilized in Liu et al. would provide greater sensitivity for detecting evolution in viral sequences. However, contrary to the suggestion in Liu et al., the detection of CTL responses against peptide epitopes in Pol would suggest that this conserved region can be considered in determining targets for vaccine design.
In the majority of these escaped responses (8 of 10), the initial CTL response against the index epitope persisted during this fall in frequency. One possibility for this is that although the level of index epitope virus decreased, some still persists to provide enough antigenic stimulation to maintain the CTL response (Fig. 4K). Another explanation would be that the initial index CTL response possesses promiscuity against evolving viral variants. These viral variants could potentially have a replicative advantage over the index epitope virus, or as a result of original antigenic sin (20), the index CTL response may be suboptimal against the variant epitopes and at the same time interfere with the maturation of naive CTL clones that would better target variant epitopes (Fig. 4L). Finally, it is possible that the index CTL response would continue to decay if trended for a longer period of time (Fig. 4J).
For the epitope KRQDILDLWVY in subject 5 (Fig. 2B) and the epitopes KRQEILDLWIY and KRQEILDLWIH in subject 4 (Fig. 4L), CTL responses against variant epitopes that subsequently developed were present at day 0, before their detection. This may indicate promiscuity of the CTL response against the index epitope rather than the development of a new response. The evolution of the circulating variants away from the index epitope may, therefore, reflect other factors that lead to a competitive advantage of viral species with these epitope sequences such as altered viral epitope processing or presentation, or improved viral replication. Although these responses in two different individuals are against the same HLA-Cw restricted epitope antigen, because the variant species noted in each subject are not similar, it is more likely that the CTL responses between these two individuals are distinct.
Because several positively screened CTL responses in subject 4 that did not show evidence of epitope escape could not be replicated during variant epitope testing (Fig. 4B–F), it is possible that this proportion of detected responses reflect a falsely positive result. This could explain the overall greater proportion of invariant targeted epitopes than previously noted (22). However, at least some of these instances may be a result of constraints placed on escape by viral fitness costs (Fig. 4I).
In studies of early HIV-1 infection, a greater response breadth, or number of HIV-1-specific CTL responses, was associated with less likelihood of escape from T cell responses within one studied individual (22). Mathematical modeling of the effect on CTL response breadth in HIV-1 infection suggested that a broader response would lead to less viral escape in early infection from a combined effect of all CTL responses in killing infected cells, balancing out the production of infected cells (38). It was also shown that increasing CTL response breadth corresponded to lower viral load in HIV-1-infected individuals; however, this association was only noted for Gag-specific antigen responses (19). Thus, although response breadth likely affects immune control in viral infection, there are other factors, such as the conservation of targeted antigen targets, that will possibly affect this relationship. Among our subjects, there was no clear association with the number of CTL responses and lower viral set point (determined by the last viral blood-level measurement in each subject). Larger number of individuals must be studied to make more definitive conclusions about the relationship between response breadth and viral set point and propensity for escape.
None of the instances of possible epitope escape in the setting of a persistent CTL index response in subject 4 were noted to have the development of a new CTL response against epitope variants. Because CD4+ T cells have been shown to be necessary to sustain (25) and promote formation of new CTL responses in chronic viral infections (18), the formation of new responses against HIV-1 epitopes may be impaired by the lack of CD4+ T cell help. Indeed, viremic control in HIV-1 infection has been associated with the presence of CD4+ T cell-dependent proliferative responses to viral antigens (17,32). The absolute CD4 count ranged from 419 to 538 cells/mm3 in this subject during the time of testing; however, functional testing of these cells was not performed. In addition, new CTL responses may also be compromised by the process of original antigenic sin (20). Although the magnitude of the response to epitope variants of the index epitopes KRQEILDLWIH and KRQEILDLWIY in subject 4 (Fig. 4J) was relatively strong, it is possible that these responses did not produce effective in vivo control of viral replication and potentially limited the formation of new CTL responses to developing variants.
Subject 4 plasma viral levels demonstrated an atypical rise after a nadir at day 12, and remained elevated throughout the time period tested. This likely reflects a poor overall CTL response not just related to escape of but also CD4+ and CD8+ T cell function.
In this study, we successfully performed an in-depth analysis of the CTL-targeted viral epitopes in six acutely HIV-1-infected individuals, and when possible, we determined the targeted minimal epitopes. The evolution of these targeted epitopes was tracked during early infection, and in one subject, we were able to comprehensively examine the relationship between these changes in viral variant epitopes and the magnitude of CTL responses against them. Our results demonstrate a period of both dynamic viral epitope variation and epitope stability under CTL pressure spanning the period of acute HIV-1 infection. This promising approach can be applied to future studies involving larger cohorts of acutely infected individuals to derive further insight into how clonal breadth, the development of new CTL responses, and other factors such as the CD4+ T cell response influence epitope escape.
Footnotes
Acknowledgments
This work was performed with grant support from the NIAID (grant numbers A1043203 and A1089398) and the AIDS Healthcare Foundation. Recombinant human interleukin-2 was provided by the NIH AIDS Reagent Repository. The University of California AIDS Institute CFAR grant provided supportive infrastructure for this project.
Author Disclosure Statement
The authors have no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.