
Abstract
Chemokines (chemotactic cytokines) are involved in a wide variety of biological processes. Following microbial infection, there is often robust chemokine signaling elicited from infected cells, which contributes to both innate and adaptive immune responses that control growth of the invading pathogen. Infection of the central nervous system (CNS) by the neuroadapted John Howard Mueller (JHM) strain of mouse hepatitis virus (JHMV) provides an excellent example of how chemokines aid in host defense as well as contribute to disease. Intracranial inoculation of the CNS of susceptible mice with JHMV results in an acute encephalomyelitis characterized by widespread dissemination of virus throughout the parenchyma. Virus-specific T cells are recruited to the CNS, and control viral replication through release of antiviral cytokines and cytolytic activity. Sterile immunity is not acquired, and virus will persist primarily in white matter tracts leading to chronic neuroinflammation and demyelination. Chemokines are expressed and contribute to defense as well as chronic disease by attracting targeted populations of leukocytes to the CNS. The T cell chemoattractant chemokine CXCL10 (interferon-inducible protein 10 kDa, IP-10) is prominently expressed in both stages of disease, and serves to attract activated T and B lymphocytes expressing CXC chemokine receptor 3 (CXCR3), the receptor for CXCL10. Functional studies that have blocked expression of either CXCL10 or CXCR3 illuminate the important role of this signaling pathway in host defense and neurodegeneration in a model of viral-induced neurologic disease.
JHMV-Induced Acute Encephalomyelitis
T
In response to intracranial (i.c.) infection of susceptible mice with JHMV, the virus rapidly spreads, infecting resident cells of the CNS (92). Within 24 h, JHMV penetrates into the parenchyma, and infects and replicates in astrocytes, oligodendrocytes, and microglia (15,92) (Fig. 1A). Brain viral titers peak between days 5 and 7 postinfection (p.i.), but decline below level of detection by plaque assay (∼100 plaque-forming unit/g tissue) between 10 and 14 days p.i. (69) (Fig. 2). Importantly, while both the innate and adaptive immune responses effectively control CNS viral replication, sterile immunity is not achieved, and viral antigen and RNA persist within the CNS (52). Viral persistence results in chronic neuroinflammation leading to an immune-mediated demyelinating disease with clinical and histologic similarities to multiple sclerosis (MS) (Fig. 3A).
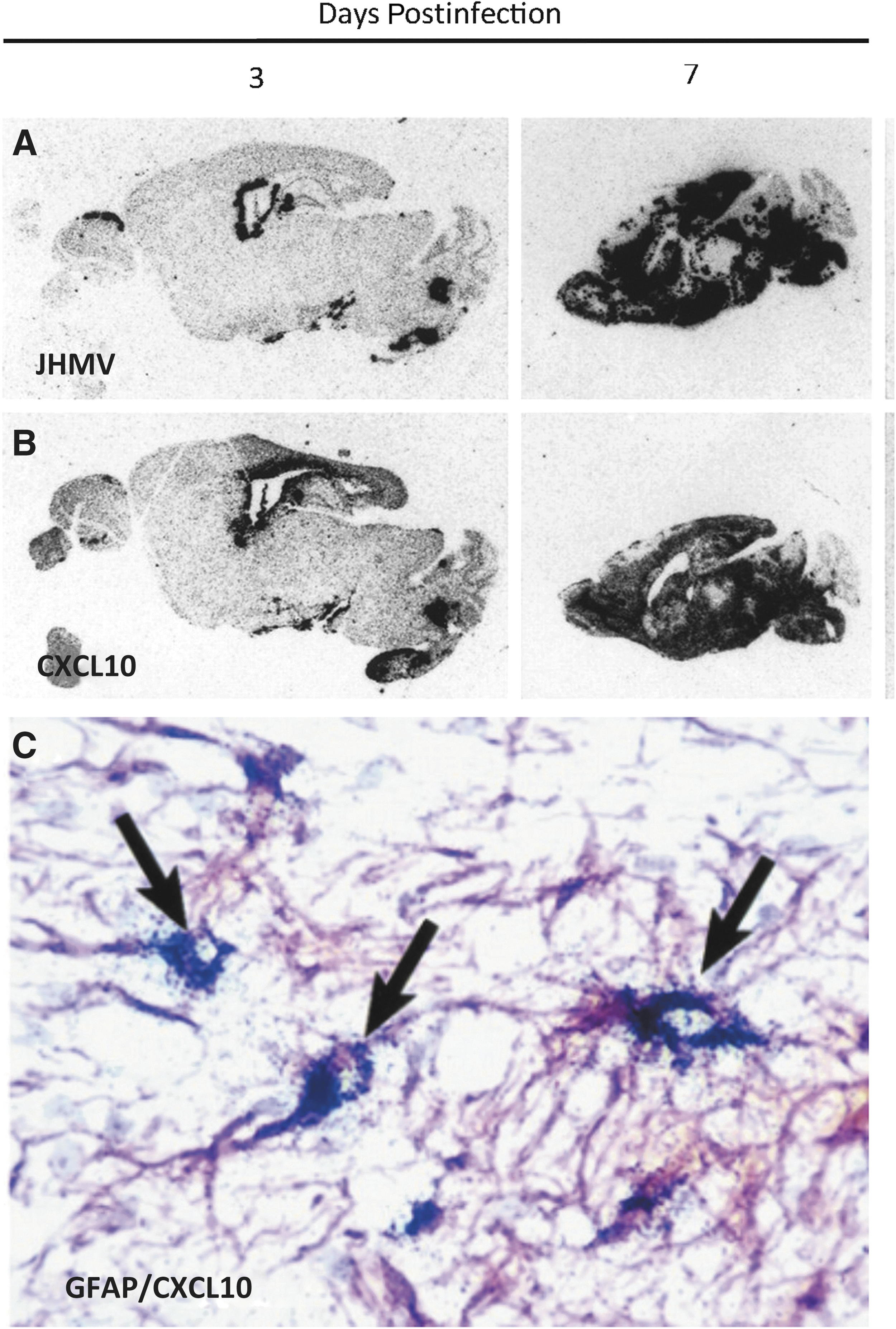
JHMV infection of the CNS induces rapid expression of CXCL10. In situ hybridization showing distribution of
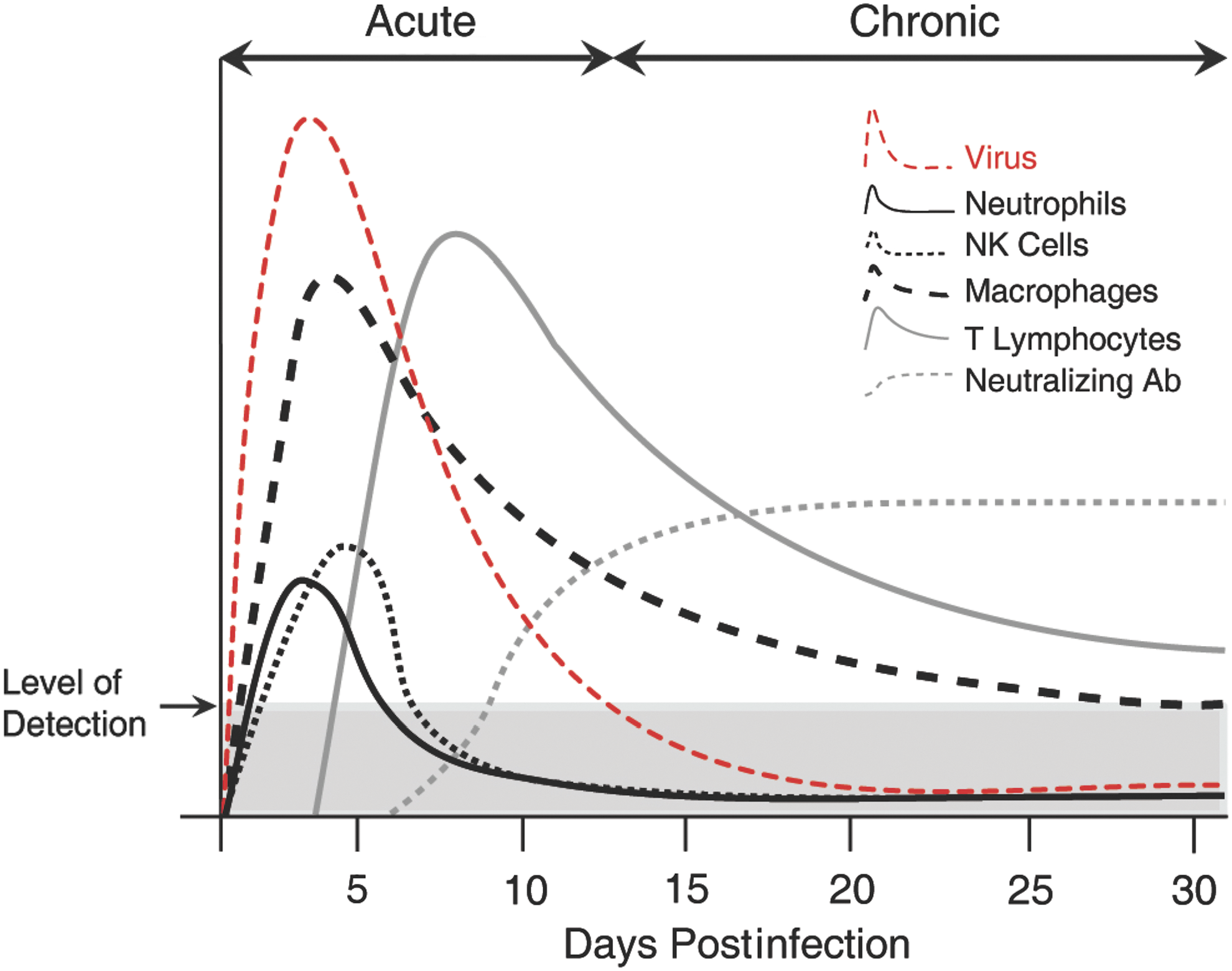
JHMV infection of the CNS invokes rapid infiltration of defined immune cell subsets. Cartoon depiction of immune response following i.c. infection of the CNS of susceptible C57BL6 with JHMV. Cellular components of the innate immune response, for example, neutrophils, macrophages, and NK cells are rapidly mobilized, and migrate to the CNS and contribute to opening the blood–brain barrier and controlling viral replication. Infiltrating CD4+ and CD8+ T cells reduce viral titers below level of detection through IFN-γ secretion and cytolytic activity. Neutralizing virus-specific antibody is required to suppress viral recrudescence during chronic disease. i.c., intracranial; NK, natural killer.
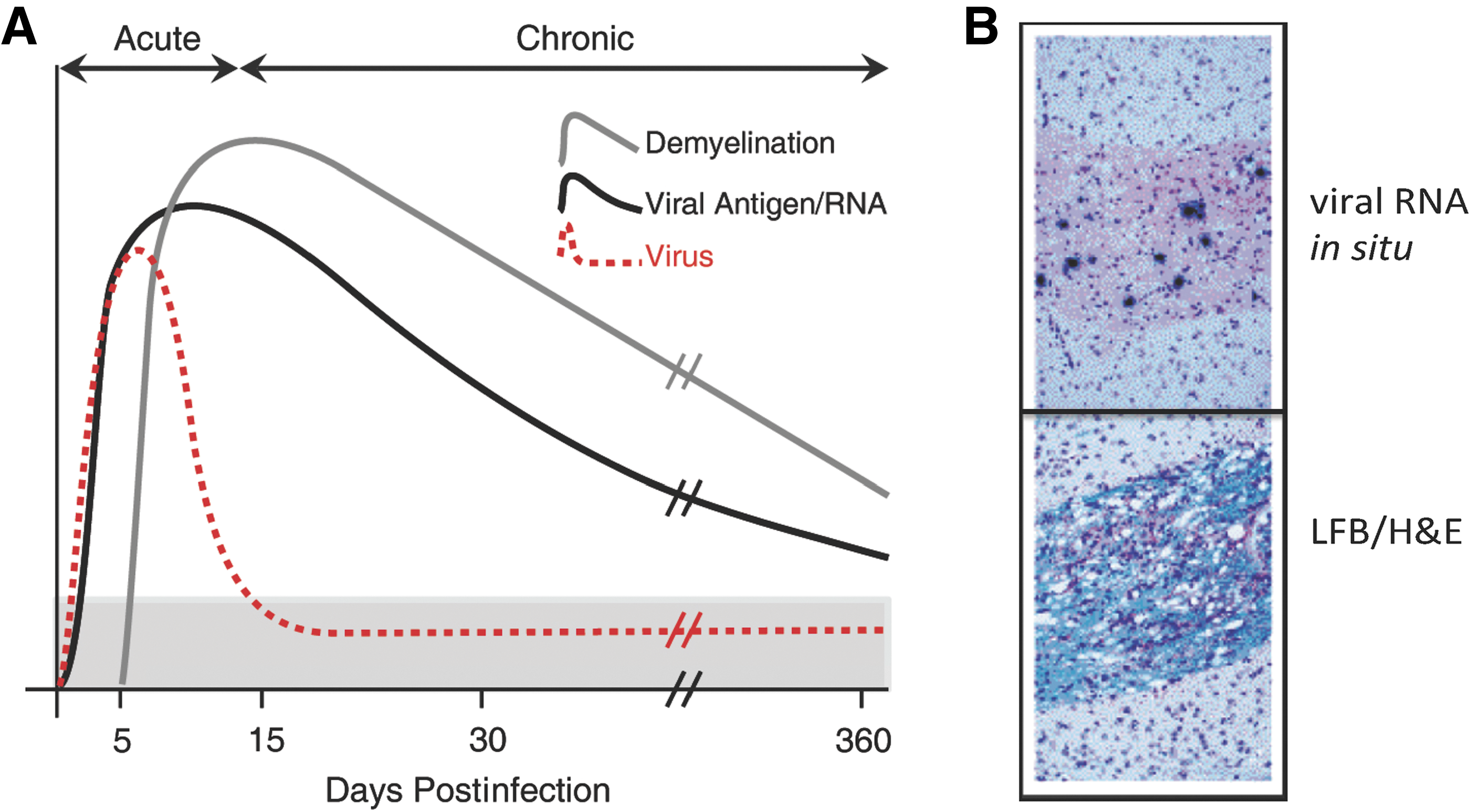
Persistent JHMV infection results in an immune-mediated demyelinating disease.
In response to JHMV infection of the CNS, there is a rapid increase in the expression of proinflammatory cytokines and chemokines along with matrix-metalloproteinases (MMPs) (5,28,104,105). Both IFN-α and IFN-β are expressed early, and elegant studies by Bergmann and colleagues (30) have implicated an important role of these cytokines in host defense by demonstrating an increase in viral spread and mortality in JHMV-infected IFNAR−/− mice. In addition, administration of type I interferons impedes viral spread throughout the CNS, further supporting an important role of these cytokines in host defense (58,75). Type I interferons have also been suggested to augment MHC class I expression arguing for a role in host defense through increased antigen presentation to T cells (1).
Neutrophils, natural killer (NK) cells, and monocyte/macrophages rapidly migrate to the CNS in response to JHMV infection (Fig. 2) (51,82,97,105). Neutrophils and monocyte/macrophages contribute to the permeabilization of the blood–brain barrier (BBB) through secretion of MMPs, and this subsequently promotes infiltration of virus-specific T cells into the CNS (28,72,101,104,105). JHMV infection of IL-15 knockout mice, which lack functional NK cells, is able to effectively control viral replication, arguing that NK cells are not required for host defense (106).
JHMV-specific CD4+ and CD8+ T cells expand to viral antigens presented within draining cervical lymph nodes, and traffic into the CNS through a permeable BBB (105). Antiviral effector mechanisms associated with viral clearance within the CNS include the elevated expression of MHC class I and MHC class II on antigen-presenting cells (APCs), after secretion of IFN-γ by both CD4+ and CD8+ T cells as well as perforin-mediated cytolysis of astrocytes and microglia by virus-specific CD8+ T cells (40,61,69). Within the context of the JHMV model, CD8+ T cell expansion and antiviral effector function are enhanced through CD4+ T cells (67). Further support for the role of CD4+ T cells in enhancing antiviral CD8+ T cell function is provided through studies in which CD4+ T cells were depleted resulting in reduced CD8+ T cell expression of IFN-γ and granzyme B combined with elevated CD8+ T cell apoptosis (67). These findings support earlier studies (80,103), demonstrating that CD4+ T cells play a crucial role in both enhancing peripheral activation of CD8+ T cells and prolonging their antiviral function within the CNS; IL-21 has been suggested to be a critical factor in controlling these specific events (67).
Oligodendrocytes infected with JHMV appear resilient to lytic effects of CD8+ T cells but are able to respond to IFN-γ secreted from virus-specific T cells and control viral replication through this mechanism (19,41,49,61). More recently, microglia have been shown to be important in host defense following JHMV infection of the CNS. Wheeler et al. (96) demonstrated increased morbidity/mortality associated with impaired antiviral effector responses by T cells following targeted deletion of microglia. These findings highlight that microglia are able to shape both innate and adaptive immune responses following infection with a neurotropic virus. With regard to B cells and their role in host defense following JHMV infection of the CNS, neutralizing JHMV-specific antibody is detected during chronic disease and is critical in preventing viral recrudescence (40,54,68,70) (Fig. 2).
More recently, Perlman and colleagues have provided important insight into the functional role of regulatory T cells (Tregs) during acute JHMV-induced CNS disease (2,102). Tregs are detected within the CNS at the same time as effector CD4+ T cells, indicating that the emergence and accumulation of both populations of cells are on a similar timeline following viral infection. Further, virus-specific Tregs express both IFN-γ and IL-10 suggesting immune regulatory capacities mediated through cytokines secreted following antigen stimulation. Indeed, virus-specific Tregs dampen proliferation of virus-specific effector CD4+ T cells, and depletion of Tregs increases mortality (2,102). These data suggest that within the context of acute JHMV-induced neurologic disease, Tregs limit immunopathological CNS disease without negatively impacting viral clearance (2).
JHMV-Induced Demyelination
Infection of susceptible mice with JHMV results in a chronic immune-mediated demyelinating disease making this an excellent and well-accepted model for the human demyelinating disease MS (6,7,36,39,50,60). Virus persists within the CNS, and in situ hybridization reveals viral RNA colocalizing with areas of demyelination in spinal cords of mice at day 35 p.i. with virus (Fig. 3A, B). A hallmark feature of JHMV infection of the CNS is characterized by viral spread into the spinal cord, with astrocytes and oligodendroglia being primary targets of infection and persistence. As a result, animals develop demyelinating lesions within the brain and spinal cord that are associated with clinical manifestations, including awkward gait and hindlimb paralysis.
Staining of JHMV-infected spinal cords with either luxol fast blue (LFB) or toluidine blue reveals demyelinating lesions concentrated within the anterior funiculus and lateral white matter columns of the spinal cord (Fig. 4A, B) (92). In addition, electron microscopic analysis of spinal cords from JHMV-infected mice reveals the extensive loss of myelin surrounding axons (Fig. 4C, D).
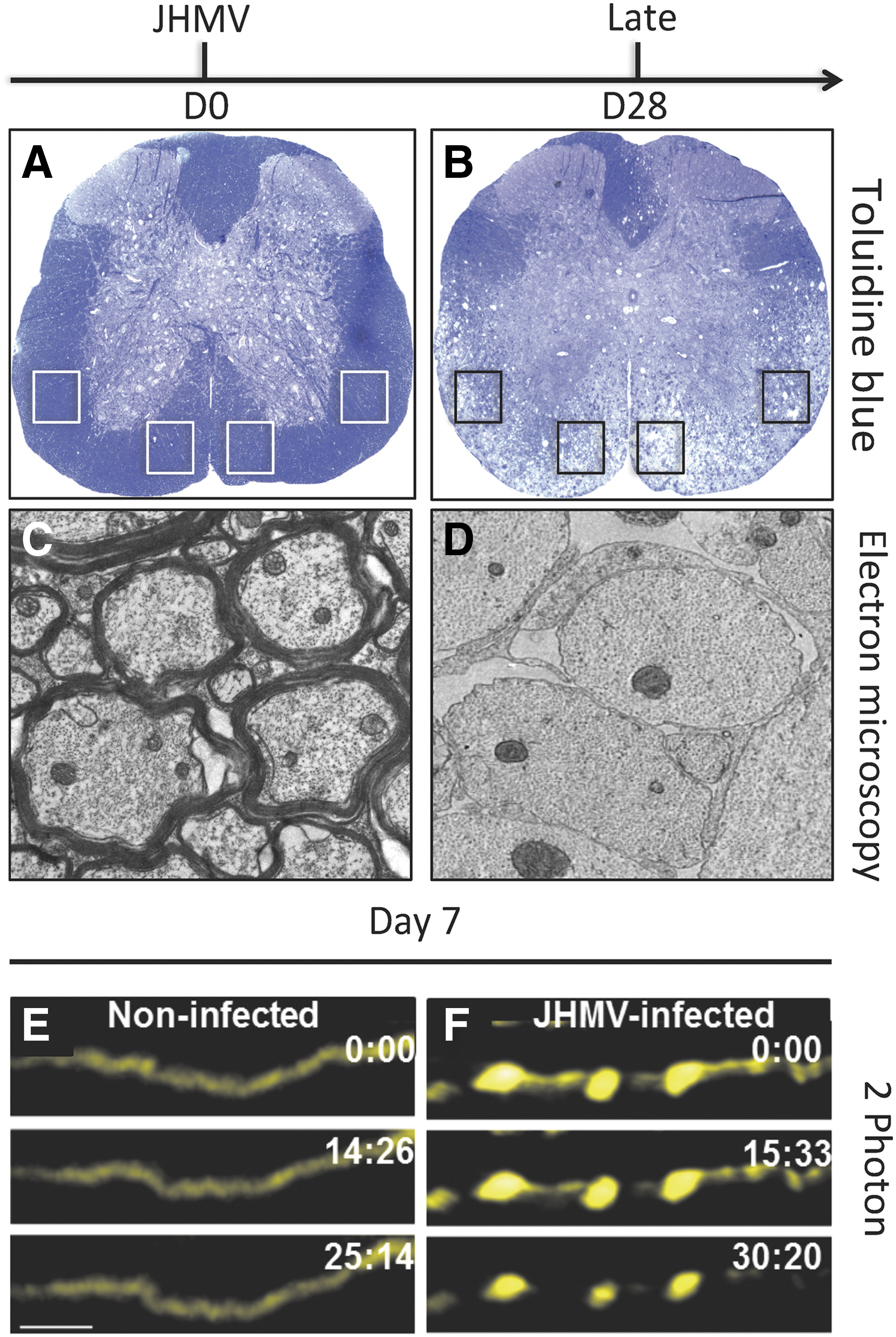
JHMV-induced demyelination and axonal damage. Toluidine blue stained spinal cord sections from
Axonopathy within the white matter tracts of the spinal cord is present as observed through the use of the SMI-32 staining or Bielschowsky's silver impregnation stain and initial observations suggested that this occurred concomitantly with demyelination, whereas axonal degeneration has been argued to precede oligodendrocyte dysregulation in MS (11,12). Indeed, our laboratory has recently employed 2-photon (2P) microscopy to visualize axonal damage in response to JHMV infection of Thy1-YFP mice in which medium-to-large caliber axons fluoresce yellow. Using this approach, we were able to detect axonal damage occurring as early as 7 days p.i. with virus, further supporting the notion that axonopathy can precede demyelination in this model (Fig. 4E, F) (21).
Current evidence suggests that demyelination in JHMV-infected mice is not the result of induction of an autoimmune response against neuroantigens, that is, epitope spreading, as has recently been reported to occur during Theiler's virus-induced demyelination (55,56). However, transfer of T cells from JHMV-infected animals into naïve recipients results in demyelination (95). More recently, Stohlman and colleagues (71) clearly demonstrated the presence of APCs capable of activating self-reactive (SR) T cells in JHMV-infected mice. SR T cell accumulation within the CNS of infected mice was shown to peak in mice persistently infected with JHMV; yet, these cells were not retained arguing for minimal pathologic function. In addition, a recent report has suggested that infection with mouse hepatitis virus strain A59 promotes activation of autoreactive T cells specific to myelin basic protein, although the contributions of these cells to demyelination remain to be fully defined (25).
Oligodendrocytes are an important viral reservoir during chronic JHMV-induced disease (15,92). Nonetheless, viral-induced lysis of oligodendrocytes is not considered a primary mechanism contributing to demyelination, as evidenced by JHMV infection of immunodeficient mice (lacking thymically-educated T and B lymphocytes), resulting in widespread viral replication within oligodendrocytes with very limited demyelination (97). Moreover, adoptive transfer of splenocytes from JHMV-immunized immunocompetent mice into immunodeficient mice infected i.c. with JHMV results in robust demyelination, implicating T cells as mediators of white matter damage (29,93,97).
Early studies from our laboratory demonstrated that JHMV-infected CD4−/− or CD8−/− mice develop demyelination demonstrating the importance of both T cell subsets in augmenting demyelination, yet CD4+ T cells may have a more important role compared with CD8+ T cells (37). CD4+ T cells secrete the chemokine CCL5, a potent chemoattractant for inflammatory macrophages, and we have shown that this is a mechanism that contributes to demyelination in JHMV-infected mice (37). IFN-γ release by CD8+ T cells also contributes to macrophage migration and accumulation within the CNS that subsequently enhance demyelination (64). Activated CD4+ T cells not specific to defined viral antigens, for example, bystander CD4+ T cells have also been shown to contribute to demyelination in JHMV-infected mice (26). Although activated CD4+ T cells are thought to amplify demyelination, in part, through recruitment of macrophages, these cells clearly exert a protective role through IFN-γ-mediated control of viral replication and/or additional undefined mechanisms (63,81). Macrophages have been shown to be important in development of demyelinating lesions within spinal cord white matter during chronic JHMV infection (16,97). Furthermore, antibody-mediated neutralization of the chemokine CCL5 or genetic ablation of its receptor Ccr5 is associated with reduced macrophage infiltration correlating with a reduction in demyelination (17,18).
Adding additional insight into how T cells contribute to either disease or defense are studies from Trandem et al. (85), showing that adoptive transfer of Tregs to JHMV-infected mice attenuates clinical disease severity, and this is associated with dampened neuroinflammation and demyelination. Clearly, T cell infiltration into the CNS of mice persistently infected with JHMV is important in the pathogenesis of disease, although a unifying mechanism(s) attributed to how these cells contribute to disease progression as well as protection remains elusive.
The Chemokine CXCL10 and JHMV-Induced Acute Encephalomyelitis
Chemokines, small (8–10 kDa) proteins expressed by almost all nucleated cell types, are divided into four subfamilies based upon the number and spacing of conserved cysteine residues present within the amino terminus of the protein. Chemokine function is controlled through often promiscuous signaling through seven transmembrane G-protein-coupled receptors. While initially characterized as important in inflammation by targeting distinct leukocyte populations, chemokines are now considered critical mediators of a variety of biological processes, including development, tissue homeostasis, and coordinated immune responses during viral infection.
The human CXCL10/IP-10 (interferon-inducible protein 10 kDa) was originally cloned and characterized following IFN-γ treatment of the human monocyte-like U937 in 1985 by Luster et al. (48). The mouse ortholog, originally dubbed cytokine response gene-2 (crg-2), was subsequently cloned and characterized in 1990 by Vanguri and Farber (89). The molecular and biochemical characterization of CXCL10 are outside the scope of this review, yet there are numerous articles detailing these specific biological aspects of this chemokine related to apoptosis (31,74), cell growth, and proliferation (38,53), as well as regulating angiostasis (99).
CXCL10 is a member of the non-ELR CXC chemokine along with CXCL9 and CXCL11, and these three chemokine ligands all bind to the surface receptor CXC chemokine receptor 3 (CXCR3) that is expressed on numerous different cell types. Binding of CXCL10 to CXCR3 expressed by cells of the immune system has been shown to influence migration/homing of macrophages, dendritic cells, NK cells, and activated T cell subsets to areas of inflammation (24,44,47). Initially described as potentially important in attracting T cells to psoriatic plaques (20), CXCL10 has subsequently been shown to be expressed in numerous human inflammatory diseases (24,32,44,47). In addition, CXCL10 is expressed in response to microbial infection, and is important in attracting targeted CXCR3-positive leukocytes to sites of infection that help control/eliminate the invading pathogen (88).
We became interested in host factors governing neuroinflammation in response to JHMV infection of the CNS. Previously, numerous cytokines had been shown to be increased in response to CNS infection, yet it was unclear whether chemokines were expressed (62). Using a RNAse protection assay (RPA) targeting chemokines, we demonstrated that transcripts encoding a number of different chemokines are rapidly synthesized in response to JHMV infection of the CNS (35). Of these, the chemokine CXCL10 is the predominant transcript detected at both acute and chronic stages of disease arguing for a potentially important role in both host defense and disease. In situ hybridization of CXCL10 transcripts revealed strict colocalization of CXCL10 messenger RNA (mRNA) transcripts with viral transcripts (Fig. 1A, B), arguing that soluble factors released from infected cells, for example, type I interferons may enhance CXCL10 expression.
We have determined that resident glial cells including astrocytes (Fig. 1C) as well as inflammatory macrophage/microglia express CXCL10 within the CNS of JHMV-infected mice (35). The early and dominant expression of CXCL10 following CNS infection by JHMV argued for a potential role as a key sentinel molecule in host defense. In support of this notion, treatment of infected mice with an anti-CXCL10-neutralizing antibody resulted in increased mortality and impaired ability to control JHMV replication that correlated with reduced levels of IFN-γ-producing T cells within the CNS (45). Therefore, these results argued that early expression of CXCL10 aided in host defense by attracting CXCR3-positive virus-specific T cells. These findings were further supported by subsequent studies employing JHMV infection of germline CXCL10−/− mice (13) that resulted in decreased entry of IFN-γ-positive T cells into the CNS and reduced ability for JHMV replication. These findings indicated that blocking CXCL10 signaling, through use of either neutralizing antibody or genetic ablation, reduced activated virus-specific T cell entry into the CNS.
Interestingly, we demonstrated through flow cytometry for staining of CXCR3 and intracellular IFN-γ following stimulation with virus-specific peptides that >90% of these virus-specific T cells expressed the CXCL10 receptor (79). However, CXCL10 neutralization selectively reduced accumulation and/or retention of virus-specific CD4+ T cells to the CNS, yet exhibited a milder effect on virus-specific CD8+ T cells (79). Furthermore, administration of anti-CXCR3 antibody to JHMV-infected mice reduced CD4+ T cell infiltration, while CD8+ T cell trafficking was not dramatically affected (78). The selective effect of anti-CXCR3 treatment on CD4+ T cells was not the result of either reduced proliferation or modulation in chemokine receptor gene expression. Therefore, CXCR3 signaling has a nonredundant role in T cell subset trafficking in response to viral infection, and argue that differential signals are required for trafficking and retention of virus-specific CD4+ and CD8+ T cells in response to JHMV CNS infection.
As an additional method to assess the importance of CXCL10 in host defense against JHMV-induced neurologic disease, we generated a recombinant virus strain of MHV capable of expressing CXCL10 (86,91). The CXCL10-expressing recombinant of MHV (MHV-CXCL10) was generated through targeted recombination using a reverse genetic approach (86). In addition, an isogenic wild-type control virus was constructed in the same manner (86). For both viruses, the exogenous gene was inserted into open reading frame (ORF)4 of the MHV-A59 parental virus (Fig. 5A). Notably, the A59 strain of MHV is capable of replicating in both the CNS and the liver following i.c. inoculation, allowing us the opportunity to explore whether the protective effects of CXCL10 are restricted to the CNS. Importantly, MHV ORF4 encodes for a nonstructural protein that is not essential for growth in tissue culture or within the mouse CNS (59,100).

MHV-CXCL10 and MHV have genetic similarity. Both viruses were generated by a recombination reaction with the thermolabile N gene deletion (designated by asterisk) mutant MHV-Alb4 and mRNA generated from a transcription reaction using plasmids that encode from upstream of gene 4 to the 3′ end of MHV-CXCL10 and MHV.
Inclusion of CXCL10 into the genome of MHV did not alter virus-specific RNA synthesis or virus-specific proteins, and resulted in secretion of CXCL10 in tissue culture (86). In addition, in vitro growth kinetics of the CXCL10-engineered virus did not alter viral replication as compared with the isogenic control virus (86). To determine whether CXCL10 expression derived from the recombinant MHV-CXCL10 resulted in enhanced protection from disease, CXCL10 −/− mice were i.c. injected with either MHV-CXCL10 or control recombinant virus, MHV. MHV infection resulted in ∼40% mortality out to day 12 p.i. (Fig. 5B). In marked contrast, 100% of mice infected with MHV-CXCL10 survived until day 12 p.i. (Fig. 5B). Our previous studies indicate that localized expression of CXCL10 within virally infected tissues is important in host defense, and peripheral expression of CXCL10 in noninfected tissues does not dramatically impact the immune response. In support of this notion, CXCL10 transcripts in CXCL10−/− mice infected with MHV-CXCL10 were selectively expressed in the CNS and liver, yet transcripts were absent in CXCL10−/− mice infected with control virus. Not surprisingly, CXCL10−/− mice infected with MHV-CXCL10 showed reduced viral titers within the brains and livers, and this correlated with increased T cell accumulation within these tissues compared with control mice. This protection from viral-induced CNS and liver disease in MHV-CXCL10-infected mice was dependent upon CXCL10 derived from the recombinant virus as treatment of anti-CXCL10 virus ameliorated these effects (Fig. 5C) (91).
Given that CXCL10 signaling has been implicated in coordinating both effector T cell generation and trafficking, we wanted to determine if CXCL10 expression following JHMV infection was important in attracting T cells into the CNS or in contributing to antiviral effector function. We have determined that MHV infection of CXCL10+/+ or CXCL10 −/− mice results in comparable levels of T cell activation and similar numbers of virus-specific CD4+ and CD8+ T cells (77). We did not detect any differences in T cell proliferation, IFN-γ secretion by virus-specific T cells, or CD8+ T cell cytolytic activity. Analysis of chemokine receptor expression on CD4+ and CD8+ T cells obtained from MHV-immunized CXCL10+/+ and CXCL10 −/− mice revealed comparable levels of CXCR3 and CCR5, which are capable of responding to ligands CXCL10 and CCL5, respectively. Adoptive transfer of splenocytes acquired from MHV-immunized CXCL10 −/− mice into MHV-infected RAG1 −/− mice resulted in T cell infiltration into the CNS, reduced viral burden, and demyelination comparable with RAG1 −/− recipients of immune CXCL10+/+ splenocytes. Collectively, these data imply that CXCL10 functions primarily as a T cell chemoattractant and does not significantly influence T cell effector response following JHMV infection (77).
While T cells clearly have an important role in controlling JHMV replication within the CNS during acute disease, antibody and B cells have a critical role in preventing viral recrudescence in persistently infected mice (54,68,70). Given the importance of antibody-secreting cells (ASCs) in suppressing re-emergence of virus, understanding how these cells migrate into the CNS is critical with regard to understanding host defense mechanisms associated with viral persistence within the CNS.
To this end, Bergmann and colleagues (65,87) have shown that ASCs express CXCR3 arguing for an important role in signaling through this receptor, and allowing these cells to migrate and accumulate within the CNS of JHMV-infected mice in which ligands CXCL9 and CXCL10 are expressed. A definitive role for CXCL10 in attracting CXCR3-positive ASCs into the CNS was confirmed through experiments in which either CXCL10−/− or CXCL9−/− were infected with JHMV and virus-specific antibody within the CNS evaluated (66). Phares et al. (66) clearly showed that ASC recruitment to the CNS of infected CXCL10−/− mice, but not CXCL9−/− mice, was dramatically impaired, thus highlighting that CXCL10 is critical for ASC recruitment. In addition to attracting ASCs to the CNS, CXCL10 was required for parenchymal entry.
CXCL10 and JHMV-Induced Demyelination
We have previously determined that CXCL10 is associated with demyelinating lesions in mice persistently infected with MHV (Fig. 6A) (35). To determine the role of CXCL10 in contributing to demyelination in mice persistently infected with JHMV, experimental animals were treated with anti-CXCL10 or control antisera beginning on day 12 p.i., which represents a time in which demyelination is established and neurologic deficits such as hindlimb paralysis are evident. Blocking CXCL10, but not CXCL9, resulted in a dramatic reduction in clinical disease severity as animals exhibited an almost complete restoration of motor skills. Importantly, clinical disease returned when we stopped anti-CXCL10 injection, further supporting an important role of CXCL10 in contributing to clinical disease (46).
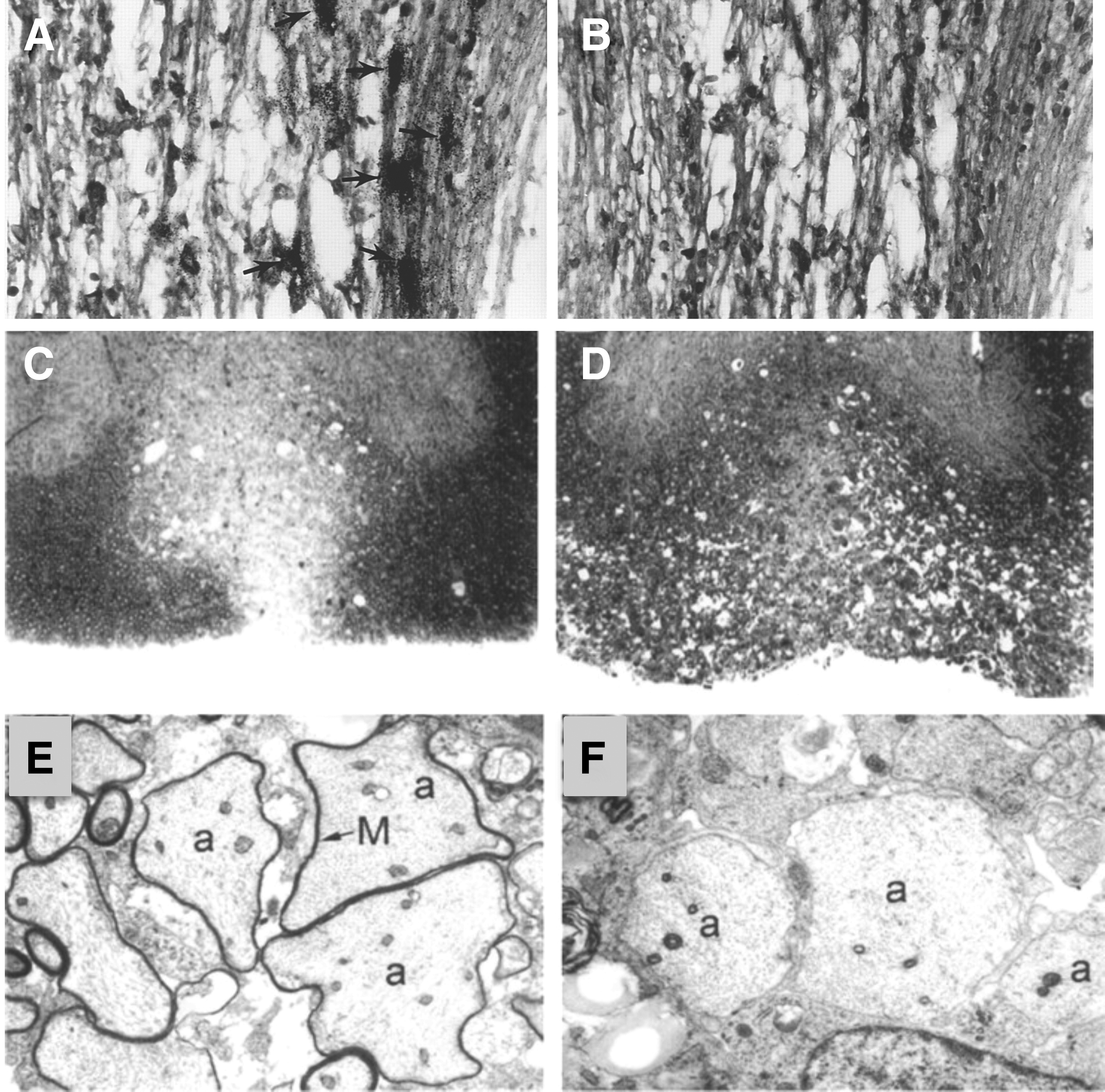
Antibody targeting of CXCL10 in mice persistently infected with JHMV reduces demyelination and increases remyelination.
We were also able to show that the muted clinical disease in anti-CXCL10-treated mice correlated with a targeted reduction in CD4+ T cells and macrophages entering the CNS as well as muted expression of IFN-γ and the macrophage chemoattractant chemokine CCL5. Furthermore, analysis of demyelination by toluidine blue staining of spinal cord sections revealed that mice treated with control sera displayed numerous inflammatory foci and robust demyelination throughout the ventral, lateral, and dorsal columns (Fig. 6D). In contrast, demyelination was limited to the ventral column in mice treated with anti-CXCL10, supporting the observation that progression of disease is impeded (Fig. 6C). Removal of anti-CXCL10 treatment correlated with a marked increase in the severity of demyelination. Evaluation of electron micrographs from anti-CXCL10-treated and control animals showed evidence of remyelination as indicated by a thin myelin sheath surrounding axons, whereas the majority of axons in control mice were entirely demyelinated (Fig. 6E, F).
The potential role of CXCL10 in contributing to demyelination in JHMV-infected mice by attracting inflammatory T cells and macrophages into the CNS was supported by additional studies, showing that demyelination was reduced in JHMV-infected CXCL10−/− mice (13) as well as in infected animals treated with anti-CXCR3 antisera (78). In both instances, the reduction in demyelination correlated with reduced CD4+ T cell and macrophage infiltration. These findings argue that blocking CXCL10 signaling results in a reduction in white matter damage by specifically inhibiting CD4+ T cells gaining access to the CNS and secreting IFN-γ that increases expression of the macrophage chemoattractant chemokine CCL5.
Interestingly, CXCL10 is increased within the cerebrospinal fluid and CNS lesions of MS patients (76), suggesting that this may be a relevant target for therapeutic intervention. Early reports using experimental autoimmune encephalomyelitis (EAE), an autoimmune-mediated neuroinflammatory disease, indicated that antibody targeting of CXCL10 blocked CD4+ T cell recruitment to the CNS, resulting in diminished clinical disease severity (14). However, subsequent studies contested these findings, and demonstrated that blocking CXCL10 either made disease worse (73) or had no effect (8). More recently, Pleasure and colleagues (57) employed a unique transgenic model in which CXCL10 was selectively ablated in astrocytes, and showed dampened disease onset that correlated with reduced CD4+ T cell entry and demyelination. Collectively, these diverse findings in different preclinical animal models of MS emphasize that the model employed may dictate experimental outcome when evaluating how CXCL10 expression influences chronic neuroinflammation and demyelination.
CXCL10 and Oligodendroglia Biology
Exposure of cultured oligodendrocyte progenitor cells (OPCs) to IFN-γ restricts proliferation and differentiation, as well as triggers apoptosis (3,4,10,22,23,27,43,90,94). Moreover, overexpression of IFN-γ within the CNS of transgenic mice results in severe behavioral deficits associated with deleterious consequences on oligodendrocytes that correlate with hypomyelination. These studies highlight the potential detrimental effect of sustained IFN-γ expression by inflammatory leukocytes infiltrating into the CNS (34,42). During chronic inflammatory diseases such as MS, OPCs/oligodendrocytes are exposed to numerous inflammatory cytokines/chemokines that create a hostile and damaging environment. Therefore, it is important to evaluate how these cells are protected from the damaging effects of IFN-γ signaling.
We have examined the mechanisms by which IFN-γ mediates apoptosis of cultured OPCs, and found that IFN-γ induces CXCL10 expression in cultured OPCs and contributes to apoptosis through a caspase-dependent mechanism (Fig. 7A, B) (84). Cultured OPCs express CXCR3, and cultures derived from either CXCR3+/+ or CXCR3 −/− mice exhibited reduced sensitivity to either IFN-γ- or CXCL10-induced apoptosis (Fig. 7C, D). Moreover, signaling through the CXC chemokine receptor 2 (CXCR2) through engagement with ligand CXCL1 restricts both IFN-γ- and CXCL10-mediated apoptosis associated with limiting cleavage of caspase 3 and increased expression of the antiapoptotic Bcl2 protein. Therefore, we would argue that in addition to contributing to demyelinating diseases through attraction of CXCR3-bearing lymphocytes, CXCL10 may have a more direct role in white matter damage through promoting oligodendrocyte loss through induction of oligodendroglia.

This increased susceptibility of OPCs to IFN-γ/CXCL10-induced apoptosis is not restricted to mice as we have also determined that treatment of human embryonic stem cell-derived OPCs with either IFN-γ or CXCL10 results in increased apoptosis through a caspase 3-mediated effect (83).
Concluding Remarks
Studies over the past 20 years from our laboratory and others have helped shape our understanding of the functional role of CXCL10 in host defense and disease in response to JHMV infection of the CNS. Using either antibody targeting or genetic silencing of CXCL10, it has been determined that early expression of CXCL10 is beneficial as it serves to attract CXCR3-positive T cells into the CNS that subsequently aid in controlling viral replication. Equally important is the demonstration that ASCs respond to CXCL10 expression in the CNS to enter the parenchyma and suppress viral replication through secretion of virus-specific antibody. Conversely, sustained expression of CXCL10 also contributes to JHMV-induced demyelination through attraction of CD4+ T cells that amplifies neuroinflammation through IFN-γ-mediated expression of other chemokines.
Importantly, subsequent studies by other investigators have demonstrated that CXCL10 is important in host defense against other neurotropic viruses, including Herpes Simplex Virus-1 (HSV-1) (98) and West Nile Virus (WNV) (33). Although much is known about CXCL10 and how it shapes inflammation in acute and chronic diseases following viral infection of the CNS, there are undoubtedly a number of additional questions that need to be addressed with regard to how the CXCL10:CXCR3 signaling pathway influences glial biology and repair in response to viral-induced neurologic disease.
Footnotes
Acknowledgments
This work was funded by the National Institutes of Health (NIH) R01NS041249 and NIH R01NS091939, as well as support from the Ray and Tye Noorda Foundation.
Author Disclosure Statement
No competing financial interests exist.