
Abstract
Viral infection in the brain can be acute or chronic, with the responses often producing foci of increasingly cytotoxic inflammation. This can lead to effects beyond the central nervous system (CNS). To stimulate discussion, this commentary addresses four questions: What drives the development of human immunodeficiency virus (HIV)-associated neurocognitive disorders, does the phenotype of macrophages in the CNS spur development of HIV encephalitis (HIVE), does continual activation of astrocytes drive the development of HIV-associated neurocognitive disorders/subclinical disease, and neuroinflammation: friend or foe? A unifying theory that connects each question is the issue of continued activation of glial cells, even in the apparent absence of simian immunodeficiency virus/HIV in the CNS. As the CNS innate immune system is distinct from the rest of the body, it is likely there could be a number of activation profiles not observed elsewhere.
Introduction
A
Inflammation in the Brain
Inflammation in the brain can be acute or chronic. Acute CNS inflammation is characterized by activation of astrocytes and microglia with increased secretion of proinflammatory cytokines, including interleukin-1β (IL-1β), tumor necrosis factor alpha (TNFα), and IL-6. This local inflammatory response begins the recruitment of leukocytes, including monocyte-derived macrophages (MDMs). Once inside the CNS, macrophages can secrete cytokines and chemokines, activating astrocytes that can then secrete more cytokines and chemokines, leading to the positive feedback loop alluded to earlier (82).
If the infectious agent is not completely controlled and eradicated, it can lead to a localized increasingly cytotoxic inflammation, which can have a cumulative effect over time and result in chronic inflammation (6,82). In chronic inflammation, the prolonged glial activation leads to tissue toxicity and neuron damage (82). To prevent this damage from spreading, reactive astrocytes can proliferate and create glial scars to block contact with healthy neurons; however, such a scar can also hinder axonal regeneration (27,58,95). That said, not all reactive astrocytes are protective, as long-term activation of astrocytes can lead to sustained innate immune activation. This will be addressed further in Question 3. In several viral infections, including simian immunodeficiency virus (SIV) and chikungunya, astrocytes can remain in an activated phenotype after immune clearance of virus, leading to chronic implications of inflammation (47,56,99).
Toll-Like Receptors
As one of the major factors of innate immunity in the CNS, Toll-like receptors (TLRs) are expressed on endothelial cells, astrocytes, microglia, and subsets of neurons. Of the 11 TLRs identified in humans, increased expression of TLR2, TLR3, and TLR9 has been reported in CNS viral infections (11 –13,38,47,56 –58). TLRs are professional molecular pattern recognition receptors that elicit the innate immune response, and later adaptive immune response, upon encountering highly specific antigens, referred to as pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (38). The release of DAMPs, as a result of cell death or tissue remodeling, can trigger activation of TLRs, assuming the correct pairing of DAMP and TLR is present at the same time (38). It is also possible that the strength of the binding between the TLR and DAMP can influence the outcome, either neuroprotective or neurodegenerative (69).
Although TLR2 is activated by interaction with bacterial products, including cell wall components and lipoprotein, TLR4 can be activated by damaged tissue markers such as soluble CD14 (sCD14) and high mobility group box-1, both of which can be shed from activated monocytes (64). Thus, TLRs contribute to brain injury through activation of microglia and astrocytes, downstream release of proinflammatory cytokines, and further recruit peripheral immune cells to the CNS. Increasing evidence is showing that once activated, astrocytes can have a prolonged increase in expression of TLR2. This is likely to have long-term effects, including macrophage recruitment and retention in the CNS, which is addressed in Question 2.
Encephalitic Viruses
Numerous viruses can cause encephalitis, including herpes simplex, cytomegalovirus (CMV), the Togaviridae and Flaviviridae, and human immunodeficiency virus (HIV). CMV has been shown to be a cause of mental retardation, cerebral palsy, and has been linked to the development of brain tumors (19,84). Togaviridae, similar to chikungunya, have neurological sequelae, including immune activation of astrocytes, even in the absence of apparent virus in tissues (47). Flaviviridae, similar to Dengue and Zika, showed hypertrophy of astrocytes in the absence of neuroinflammation and vascular leakage, indicating activation of astrocytes that continued after viral control (57).
Perhaps the most widely studied virus associated with encephalitis is HIV. HIV and the parent virus SIV subvert the innate and adaptive immune responses, in part, through infection and reprogramming of myeloid cells (49,82). Because of this, HIV and its model, SIV, are the focus of this commentary.
Studies of HIV infection of CD4+ T cell subpopulations have become increasingly popular (22). However, too often analyses of other cell types are excluded. Once CD4+ T cells become infected, they experience cytopathic effects and often die, which is why a CD4+ latent reservoir model is losing popularity. Viral outgrowth analyses have shown that there are greater number of infected monocytes and MDMs than CD4+ T cells in blood, bronchoalveolar lavage, lungs, spleen, and brain (3). It has almost become forgotten that HIV is a lentivirus and, therefore, infects monocytes and traffics to the CNS within a Trojan horse (21,92). Beyond this, there is much debate, fueled by several studies that suggest, but without proving, a lesser role of macrophages and other cell types. This commentary seeks to pose some questions that some may see as controversial.
What drives the development of HIV-associated neurocognitive disorders (HANDs)?
Does the phenotype of macrophages in the CNS spur development of HIV encephalitis (HIVE)?
Does continual activation of astrocytes drive the development of HANDs/subclinical disease?
Neuroinflammation: friend or foe?
Q1: What Drives the Development of HANDs?
HANDs are the collective term for HIV-related neurological impairment. HANDs are broken down into asymptomatic, referred to as asymptomatic neurocognitive impairment (ANI), and symptomatic, which is further broken down into the classifications mild neurocognitive disorder/impairment (MND/I) and HIV-associated dementia (HAD). Pathologically, HIVE has been defined by perivascular leukocytic infiltrates, perivascular cuffing, and gliosis, but the unique diagnostic criteria are the presence of multinucleated giant cells (MNGCs) in the brain (80). MNGCs are giant cells created by the cell–cell fusion of macrophages in response to chronic inflammation, as is seen in chronic HIV infections (61,63).
Despite the advent of combination antiretroviral therapies (cARTs) and the resulting suppression of viral replication, the prevalence of HIV-associated neurocognitive impairment has increased (5), most likely due to ongoing neuroinflammation. The 2010 CHARTER study found that >52% of HIV-infected individuals had a neurocognitive disorder, with 7% diagnosed with HAD, 12% diagnosed with MND/I, and 33% with ANI (5,41). Patients with HAND and HIVE experience worse adherence to treatment, lower quality of life, and a higher rate of mortality than patients without it (5,65,66). Another study found that 25–30% of untreated adults with HIV-1 and 15% of patients treated with cART had HAND (77). Many hypotheses have been proposed, such as a legacy effect, which suggests damage was already done before the initiation of therapy (78), cART neurotoxicity, and the inability of cART to cross the blood–brain barrier (BBB) (5). It is possible that this damage is due to chronic activation of astrocytes and other glial cells of the brain, which are chronically activated in SIV/HIV infection and can persist in the absence of virus in CNS (56). We will return to this idea in Question 3.
Monocyte invasion is linked to the development and pathogenesis of HIVE (40,62), and increased monocyte turnover predicts the progression of HIV infection to AIDS, and is also correlated with the severity of HIVE (10). There are three subsets of monocytes, which have since been termed classical, nonclassical, and intermediate monocytes. Classical monocytes are characterized by high CD14 expression, low-to-no CD16 expression, and moderate CD64 expression and comprise about 90% of the circulating monocytes in blood. Intermediate and nonclassical monocytes make up the rest of the monocytes circulating and differ in that they express CD16. Intermediate monocytes are distinguished by high CD14 and CD64 expression, whereas nonclassical monocytes have low CD14 expression and no CD64 expression (2). CD16+ monocytes are selectively infected with HIV, are preferentially attracted to the endothelial receptors of the BBB, and have increased junctional proteins such as JAM-A, ALCAM, and PECAM-1 to mediate the diapedesis (60,87,88,91). CD16+ monocytes have also been shown to promote higher viral replication than CD16− monocytes when they differentiate into macrophages and interact with T cells (2).
There are two overlapping theories for how monocytes enter the CNS, the “Trojan horse” and the “Trojan herd.” The monocyte “Trojan horse” model theorizes that an infected monocyte traffics across the BBB during normal immune surveillance early in infection [usually around 10–14 days postinfection (55)]. Once this infected monocyte enters the CNS, it becomes a productively infected macrophage, inducing more (potentially infected) monocytes to cross the BBB, forming perivascular cuffs of cells and triggering limited breakdown of the BBB, including the choroid plexus (26,70). It is believed these infected monocytes cross the BBB and differentiate into MDM and infect CNS resident macro- and microglia and lead to viral latency (49,54). Recently, the “late invasion” or “Trojan herd” model was proposed, where changes in the circulating monocyte phenotype described earlier can lead to increased cycles of invasion of monocytes in late-stage infection (30,34).
Macrophages and microglia are the primary targets of HIV infection in the CNS (18,97). Since both activated microglia and MDMs secrete proinflammatory cytokines, it has been debated over which drove SIV infection of the brain; however, in vivo and in vitro (74) studies suggest it is more likely that infiltrating macrophages drive the microglial response to SIV infection. These microglia activated by infiltrating MDMs had increased levels of monocyte chemoattractant protein-1 (MCP-1/CCL2), granulocyte–macrophage colony stimulating factor, and TNFα, regardless of whether the macrophages were SIV infected or not, although there were increased levels of IL-6, IL-8, and vascular endothelial growth factor when SIV-infected macrophages were introduced (74).
Q2: Does the Phenotype of Macrophages in the CNS Spur Development of HIVE?
SIV encephalitis is only seen in chronic infection with macrotrophic strains of SIV, providing further evidence of macrophage involvement in AIDS disease progression (83). At least three types of macrophages have been identified within the brain: microglia, perivascular macrophages, and choroid plexus macrophages (36,50). It was once thought that microglia populated the brain early in utero with very little turnover during the life span (94). Recent evidence using selective depletion of microglia (24,45), however, shows the presence of a population of microglia progenitor cells.
Perivascular macrophages have a comparatively short turnover: within 14 weeks, based on labeling studies (4). Perivascular macrophages were identified as a major cell productively infected during acute and terminal SIV infection and form the characteristic macrophage cuffs and MNGCs that are the defining pathological characteristic of HIVE (93).
Within the choroid plexus, there are two distinct populations of macrophages: choroid plexus macrophages are believed to be long-term tissue established macrophages, whereas perivascular macrophages, which lie along the fenestrated capillaries inside the choroid plexus, are the short-lived MDMs (36).
Changes to monocyte and macrophage phenotype and differentiation can lead to the development of HANDs (20,29). The dogma used to be that there were two phenotypes of CNS macrophages, proinflammatory M1, and anti-inflammatory M2, although recent studies have expanded the macrophage polarization to also include MØ and M4. M1 and M2 are the most widely characterized, with increasing number of subsets for M2 phenotypes proposed, including M2a, M2b, and M2c. M1 is believed to be classically activated and proinflammatory (triggering Th1 responses and secretion of IL-1, IL-12, TNFα, and ROS), whereas M2 is alternatively activated and anti-inflammatory (triggering Th2 responses and secretion of IL-10) (17,48). Ourselves and others have shown the presence of TNFα-producing macrophages in macaques with SIVE (67), yet M1 polarization may inhibit HIV from even integrating into macrophages (81). The time of monocyte recruitment, early or late in infection, can polarize the monocytes to either M1 or M2, respectively (37).
The type, degree, and length of macrophage activation can drive neuroprotection or neurodegeneration (1), leading to apparently contradictory phenotypes. For example, a receptor for haptoglobin–hemoglobin, CD163, has been described as a marker of M2 macrophages (25,31). Conversely, upregulation of CD163 on monocytes may follow activation of TLR2 (89). M2-polarized perivascular macrophages, double positive for CD163 and CD16 (which we described earlier as being preferentially infected with HIV), can be found in the CNS early in infection (7,25), and correlated with plasma viral load and negatively correlated with CD4+ T cells (10,31). However, CD163 is not an exclusive marker for perivascular macrophages; it can also identify activated microglia and choroid plexus macrophages (7,53). To complicate the issue further, CD163 is shed from monocytes/macrophages after exposure to TNFα, soluble CD163 is elevated in chronic HIV infection (9,25) and correlates with the percentage of intermediate and nonclassical monocytes in HIV infection (9). Thus, it is possible that M1 polarization, traditionally considered proinflammatory, actually prevents HIV replication, whereas the anti-inflammatory M2 phenotype may be more permissive to HIV replication and disease progression (68).
CD206 was originally thought of as a selective marker for perivascular macrophages, linked to an M2 phenotype. A phenotypic shift from CD206+ to CD206− perivascular macrophages has also been observed in SIV-infected rhesus macaques, indicating that the virus might cause changes in macrophage phenotype (46). Increased migration and proliferation of macrophages [a hallmark of M1 polarization (48)] lead to their accumulation and ultimately the formation of MNGCs, which are usually positive for viral RNA (39). As productive viral infection in macrophages potentially indicates an M2 phenotype, then the proliferative M1 neuroprotective response might be futile (28,46). It is also possible that the accumulation of macrophages could contribute to the damage of HIVE by polarizing more M2 macrophages from infiltrating myeloid cells (96).
Q3: Does Continual Activation of Astrocytes Drive the Development of HANDs/Subclinical Disease?
Another potential source of increased monocyte invasion in both acute and chronic HIV infection is activated astrocytes. For too long, astrocytes were considered support cells maintaining neuron health and contributing to BBB integrity (85). A protective role was acknowledged by protecting neurons from inflammation by helping repair the BBB and preserving surrounding tissue through the formation of glial scars (32,85), as discussed earlier. Indeed, there may be no direct counterpart for astrocytes in the peripheral immune system (72). However, astrocytes also play a role in the inflammatory damage linked to HIVE. Although astrocytes have not been reported to be productively infected in adults, they are postulated to be a latent reservoir even under ART. Furthermore, once astrocytes are infected during acute infection, they do not return to normal function as evidenced by long-term dysregulation of TLRs (56). Astrocytes are capable of being infected as they express both the CXCR4 and CCR5 coreceptors needed in HIV infection with restricted infection of astrocytes reported in neonatal rhesus macaques (59,90), although this may require specific tropism of viral strains (59).
Astrocyte activation could drive the increased monocyte turnover seen in HIV infection and HIVE. Astrocytes can be activated through binding transactivator of transcription (Tat) or cytokines secreted by neighboring microglia/MDMs. This leads to increased expression of the adhesion molecules intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 (86). These adhesion molecules then facilitate increased monocyte migration into the brain through chemotaxis and haptotaxis (35,52). Increased proinflammatory cytokines have been observed in brain tissue after SIV/HIV infection, including CCL2/MCP-1 and MCP-3/CCL7 (73,79,98). Although MCP-1/CCL2 recruits both monocytes and T cells, CD4+ T cells are not found in large quantities in the CNS in HIV infection (16,51). It is interesting to note that astrocytic supernatant had increased CCL7 and induced preferential migration of MDMs versus T cells (73). This could explain why monocytes, rather than CD4+ T cells, are preferentially selected to the CNS during HIV infection.
Under resting conditions, astrocytes express several TLRs that can bind DAMPs and PAMPs, activating an immune response (71). In HIV infection of the CNS, TLR2 is upregulated in astrocytes, which has been linked to sustained inflammation due to the continuous secretion of proinflammatory cytokines, and sustained TLR2 expression, (8,23,42,56) even when no virus is evident in the CNS (56). This is combined with decreased TLR9 expression (23). It is easy to imagine how HIV hijacking of the normal innate immune function of astrocytes would have long-standing effects (Fig. 1).
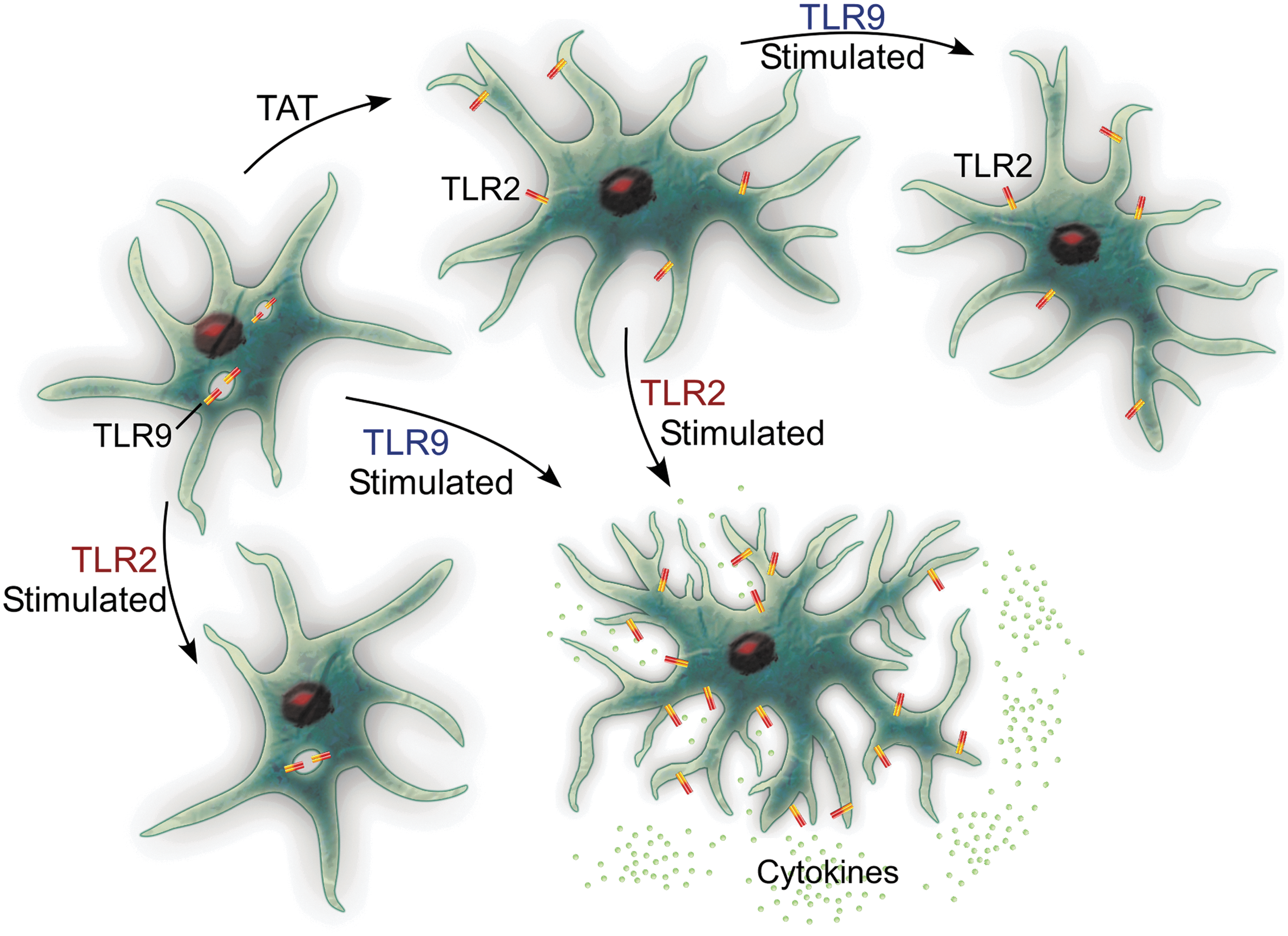
Under normal healthy conditions, astrocytes express high levels of TLR9 (generally thought of as antiviral) and low levels of TLR2 (antibacterial). When stimulated through TLR2, cytokine secretion is low, whereas exposure to TLR9 agonists produces a higher secretion of cytokines. When astrocytes are exposed to HIV proteins, including Tat, there is a phenotypic switch, both in vitro and in vivo. This means that if the astrocytes are then stimulated with TLR9 agonists, they fail to produce cytokines, yet when stimulated through TLR2, they produce a cytokine response. This implies that Tat protein hijacks the glia and creates increased activation through TLR2, while limiting the response to HIV viral antigens through TLR9. HIV, human immunodeficiency virus; TLR, Toll-like receptor.
Under normal conditions, astrocytes have high TLR9 (considered antiviral) and low TLR2 (considered antibacterial) (23,42,43). Thus, stimulation with a TLR2 agonist results in fairly low levels of cytokines, but TLR9 binding would be anticipated to induce higher secretion of cytokines. Incubation with Tat protein induces decreased expression of TLR9 (23) and increased TLR2; stimulation with TLR2 agonists induces increased cytokines (23,44). Thus, Tat effectively hides astrocytes from the very infectious agent they are stimulated by, making the astrocytes refractory to stimulation through viral antigens! This could provide an avenue through which astrocytes remain continually primed for activation, just not to the very virus that has primed them.
Q4: Neuroinflammation: Friend or Foe?
Arguably, the damage associated with HANDs is not caused by the virus, per se, but rather by the double-edged sword that comprises the immunological response(s) within the CNS. Obviously, inflammation is needed to control infection and the damage caused by it; however, HIV may have subverted this system, allowing for long-term and progressive damage to the CNS. The HIV proteins glycoprotein 120 (gp120), Tat, and negative regulatory factor (Nef) may do more damage than the actual HIV infection of the CNS cells.
HIV infection and damage of the brain is a multifaceted positive and negative feedback system composed of monocyte/macrophage, microglial, and astrocytic components. As we have shown earlier, classically activated proinflammatory M1-polarized macrophages (including microglia) are refractory to HIV replication; however, M2 macrophages, which should be resolving neuroinflammation, allow HIV to replicate (Q2). The result is an endless cycle of resolving and relapsing activation of microglia and astrocytes (Q3), waves of infiltrating monocytes (Q1), leading to chronic damage.
Once neuroinflammation is resolved, it is possible that the damage might still exist. A difficulty in assessing the presence of active inflammation in animal models is the histology at necropsy. If MNGCs are not identified on the hematoxylin and eosin slide, does that mean the animal never experienced neuroinflammation? It is not even known whether the brain returns to normal once MNGCs are present or whether this is a “point of no return?” Are other nonimmune cells of the brain damaged permanently? Pathologists are limited by the timing of necropsies, and H&Es do not always identify activation of nonimmune cells, including astrocytes and endothelium.
Some potential solutions moving forward would be to target numerous aspects of CNS infection, thus preventing initiation of the positive feedback loop. The best solution remains to prevent HIV prophylactically, and the second should be early and quick detection that allows for treatment with cART with high neuropenetrance. Alternative solutions under active investigation include blocking trafficking of infected monocytes into the CNS (14,15). Another alternative therapy that is gaining popularity is the use of tetrahydrocannabinol (THC). Levels of circulating CD16+hi monocytes are lower in HIV+THC+ patients than in untreated HIV+ patients, possibly by preventing the CD16− to CD16+ transition (75). THC also blocks the migration of microglial-like cells to HIV tat (33). Further studies are needed to elucidate the positive feedback loop in HIV infection, as well as treatment options to target different parts of the positive feedback loop to prevent rampant chronic damaging cellular activation and subsequent neuroinflammation.
Footnotes
Acknowledgments
This commentary was supported by grants OD011104 (formerly RR00164), NS104016, and MH113517 from the National Institutes of Health to the Tulane National Primate Research Center.
Author Disclosure Statement
No competing financial interests exist.