
Abstract
The mechanistic mammalian target of rapamycin (mTOR) plays a crucial role in response to many major cellular processes, including cellular metabolism, proliferation, and autophagy. Both mTOR and autophagy are suggested to be involved in the viral infection. However, little is known about the role of mTOR and autophagy in human endothelial cell infected with dengue virus-2 (DENV-2), this study is to investigate the role of mTOR and autophagy in human umbilical vein endothelial cells (HUVECs) infected with DENV-2 and related regulatory mechanisms. HUVECs were cultured in epithelial cell medium. A series of experiments involving immunohistochemistry, TCID50 method, real-time PCR, western blot, and laser confocal were performed in this study. The cell line was identified as HUVEC by the expression of cell factor VIII. The expression level of DENV-2 mRNA increased and showed an upward trend. Compared with the control group, the fluorescence of autophagy-labeled protein LC3B and lysosome-labeled protein lysosome-associated membrane protein 1 (LAMP1) in the cytoplasm of HUVEC induced by rapamycin was observed, and intensity was significantly enhanced under confocal laser scanning microscope, after fluorescence synthesis, the fluorescence of autophagy-labeled protein LC3B and lysosome-labeled protein LAMP1 overlaps were reduced. The intensity of fluorescence of autophagy-labeled protein LC3B and lysosome-labeled protein LAMP1 increased in 1 × 104 TCID50 DENV-2 infection group, after fluorescence synthesis, fluorescence of autophagy-labeled protein LC3B, lysosome-labeled protein LAMP1, and DEN2 NS1 overlapped. Compared with the control group, the phosphorylation level of mTOR, Atg13, and p-ULK1 in DENV-2-infected group or Rapa treatment group decreased significantly (p < 0.05), and the level of LC3-II increased significantly (p < 0.05). These results suggest that DENV-2 induces autophagy in HUVECs through mTOR signaling molecule.
Background
The dengue viruses (DENV) are members of the Flavivirus genus of Flaviviridae family, which are positive-strand RNA viruses. There are four distinct dengue virus (DENV) serotypes that share antigenic relationships (DENV-1, DENV-2, DENV-3, and DENV-4) (32,34). DENV is transmitted by insects such as Aedes aegypti and Aedes albopictus, causing dengue fever (DF), dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) with high mortality (9,28). DENV infection is widespread in tropical and subtropical regions. WHO estimates that 2.5 billion people are at risk of infection of DENV in worldwide each year, the impact of DENV on human health has become more and more serious in the past decades. DENV infection has become a major public health problem facing the world (21,26). DHF and DSS are serious clinical types of DENV infection, of which main pathological features are swelling, increased permeability and hemorrhage of vascular endothelial cells (9). The pathogenesis of DHF and DSS has not been elucidated. In this study, the mechanism of DENV infection was studied via cultured human umbilical vein endothelial cells (HUVECs) in vitro.
Autophagy was referring to the degradation of intracellular substances such as damaged proteins and organelles to maintain cell homeostasis, to achieve the needs of metabolic of the cell itself and the renewal of certain organelles (20,31). Autophagy is crucial for maintaining the renewal of cell components and maintaining the vigorous physiological state, autophagy is divided into three types, microautophagy, chaperone-mediated autophagy and macroautophagy (17). Macrophage is the main pathway of degradation of cytoplasmic components, which mediates the formation of autophagosomes, and then fuses with lysosomes to achieve the degradation of contents (25). Autophagy is a double membrane vesicle that reclaims and degrades proteins and organelles through cup-shaped extensions (35). Whether autophagy plays a positive or negative role in the physiological and pathological processes of organisms has not been fully elucidated, especially in the study of viruses. Recent studies have shown that autophagy plays a dual role in viral infection and viral replication. Cells can use autophagy to resist viral infection and can also use autophagy to protect viruses from inhibition and elimination of cells (23). In recent years, more and more attention has been paid to the regulatory role of mammalian target of rapamycin protein (mTOR) in various physiological and pathological processes. The mTOR signaling pathway can be used by organisms and cells to regulate various downstream signaling pathways, thus regulating gene transcription, protein translation, liposome synthesis, energy metabolism, and autophagy (2). In many related pathways, the autophagy process of mTOR and its downstream play a central regulatory role in maintaining the growth and metabolism of the body and cell (2). mTOR is an atypical serine/threonine protein kinase, including two complexes, namely mTORCl and mTORC2 (33). Due to the difference of rapamycin sensitivity between the two complexes, most of the current studies on mTORCl have focused on the complex. Studies have shown that autophagy has certain effects on some vascular diseases (22). Clinical studies have shown that vascular diseases can induce autophagy in viruses (3). The mechanism of autophagy on virus infection is not yet fully understood. Recent studies have shown that signaling pathways regulating autophagy may be a new strategy for preventing or treating human diseases, autophagy controls the process of viral infection by regulating the innate immune response (13). However, the regulatory mechanism of mTOR and autophagy in human umbilical vein epithelial cells in DENV infection is still not very clear. The aim of this study is to investigate the relationship between autophagy and DENV-2 infection in human umbilical vein epithelial cells by the mechanism of autophagy.
Materials and Methods
Virus infection
HUVECs grown to a single layer were infected with DENV-2 (1 × 102, 1 × 103 and 1 × 104 TCID50). Serum-free epithelial cell medium cultures (1% endothelial cell growth supplement (ECGS) and 1% penicillin/streptomycin (P/S)) were used for virus formulation. Viral infection of HUVECs was carried out in an incubator at 37°C with 5% CO2 for 2 h. Unattached viruses were then washed off using Hank medium, and the cells were incubated and maintained in culture medium (2% fetal bovine serum [FBS], 1% ECGS,1% P/S). Samples were collected at 4, 8, 12, 24, 36, and 48 h after infection.
Main reagents
Reagents used were medium 199/EBSS (HyClone, Japan); FBS (GIBCO); ECGS (ScienCell); HUVEC (ScienCell Cat. No. 8000); DAB Reagent Kit (Zhongshan Jinqiao, China); Factor VIII-related antigen F rabbit polyclonal antibody (Zhong shan Jin qiao, China); Platinum SYBR Green qPCR (Invitrogen); DNase I amplification grade (Invitrogen); rapamycin (Blue sky, China); bafilomycin A1 (SIGMA, Japan); anti-LC3B antibody (Abcam, ab48394); anti-LAMP1 antibody (H4A3)-lysosome (Abcam, ab25630); anti-dengue virus NS1 glycoprotein antibody (Abcam, ab41623); goat anti-mouse IgG, Dylight™ 550 (Pierce, 84540); 4′,6-diamidino-2-phenylindole (SIGMA, Japan); antiphospho-(Ser2448)-mTOR antibody (Cell Signaling Technology); ULK1 (A705) antibody (Cell Signaling Technology); antiphospho-(S757)-ULK1 (Cell Signaling Technology); Atg13 (E1Y9V) (Cell Signaling Technology); phospho-Atg13 (Ser355) antibody (Affinity Biosciences, OH).
Immunohistochemistry
1 × 105 HUVEC cells per well were cultured at 37°C and 5% CO2 for 24 h in six-well plates. Phosphate-buffered saline (PBS) washing three times, ice methanol fixed for 20 min, 0.3% Triton X-100 broken membrane for 10 min, 3% H2O2 room temperature sealing for 15 min, 0.03% Triton X-100 wash three times with 2 min each time. The monoclonal antibody labeled with HRP (1:50) was diluted and slowly added into the center of the glass slide, 200 μL/well, and washed at 4°C overnight, 0.03% Triton X-100 for three times with 2 min each time, and then the color was developed by dab. The color development time was controlled under microscope.
Real-time PCR relative quantification method to detect the level of DENV-2 mRNA in DENV-2 infected HUVEC
Cells seeded in six-well plates as above described were infected with DENV-2 virus (1 × 102, 1 × 103 and 1 × 104 TCID50) for 24 h. The cells were collected for RNA extraction. The total RNA was extracted by TRIzol method and the concentration of RNA was detected by ONE DROP, according to the manufacturer's instructions, and the first strand cDNA was obtained by reverse transcription with the Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) using 1.0 μg of total RNA as a template. For each time point, three replicate extractions of RNA samples were performed. Real-time fluorescence quantitative PCR was performed using cDNA as template. The Ct value was recorded, the average value was calculated. According to Ct = DENV-2 treatment group (Ct target gene -CtGAPDH) –CtGAPDH-negative control group (Ct control gene -CtGAPDH), 2-ΔΔCt was calculated, and 2-ΔΔCt was used for statistical analysis.
Western blotting
Cell samples were prepared at different times after DENV-2 infection and drug treatment, SDS-PAGE electrophoresis of extracted proteins was carried out, and the proteins were then transferred to a polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was blocked for 2 h and then incubated overnight at 4°C with primary antibodies. After washing three times with Tris-buffered saline–Tween-20 (TBST), the membranes were incubated with secondary antibodies at 37°C for l h and then washed three times with TBST. The ECL luminescent substrate was added to the PVDF membranes and the color was developed. The densities of the immunoreactive protein bands were analyzed using the Quantity One software (Bio-Rad, Hercules, CA). The ratios of LC3B-II, p-mTOR, Atg13, p-Atg13 (Ser355), p-ULK1 (Ser757), and ULK1 to β-actin were calculated.
Laser confocal microscopy
HUVECs were cultured at 1 × 104 cells in each well in confocal culture dishes for 2 days and then treated with bafilomycin A1 (20 μM) for 24 h, or infected with DENV-2 (104 TCID50/mL) for 24 h. The sample was then fixed with paraformaldehyde for 10 min. Then, 2% Triton X-100 (Solarbio) was used to permeabilize the cells for 10 min. The sample was washed three times with PBS. After the cells were blocked with 5% BSA (Solarbio) g for 30 min, they were incubated with primary antibodies (anti-[1:100 rabbit anti-LC3B, 1:10 mouse anti-LAMP1, 1:5 mouse anti-NS1], fluorescent secondary antibodies [1:10 FITC-labeled anti-rabbit, 1:200 633-labeled anti-mouse, and 1:200 550-labeled anti-mouse]) were incubated at 37°C for 1 h. Finally, the cells were incubated with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI, Solarbio) at 37°C for 10 min, and then observed under a laser confocal microscope.
Statistical analysis
Data are expressed as mean ± standard deviation of at least three independent experiments and were analyzed using Prism software (GraphPad, Inc., La Jolla, CA). Differences between two groups were assessed using unpaired two-tailed Student t-test. Data involving more than two groups were assessed using ANOVA. p values less than 0.05 were considered significant (*p < 0.05; **p < 0.01 and ***p < 0.001).
Results
Identification of HUVEC (IHC)
HUVEC factor VIII-related antigen was detected by IHC. Compared with the negative control (Fig. 1A), obvious brown-yellow particles were observed in the cytoplasm of HUVEC (Fig. 1B), which was confirmed as endothelial cells.
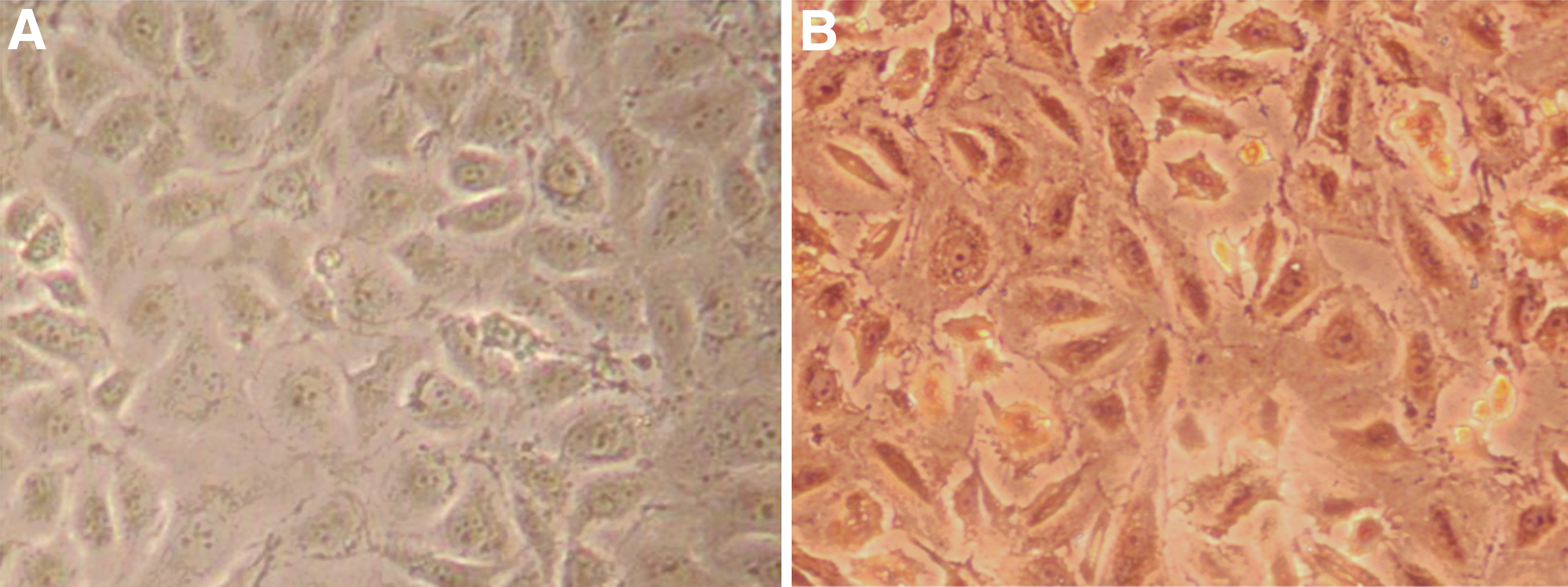
Immunohistochemistry of factor VIII in HUVEC (200 × ).
Determination of virulence of C6/36 cells by DENV-2
Typical cytopathic changes such as swelling, fusion, and vacuolization of C6/36 cells occurred from 5 to 7 days after DENV-2 inoculation. As shown in Table 1, the TCID50 was calculated to be 10−5.6 according to the Reed–Muench method.
Titration of DENV-2 Infected (TCID50 Calculation)
Real-time PCR relative quantitative detection of DENV-2 mRNA level in HUVEC infected with DENV-2
As shown in Figure 2A, real-time fluorescence quantitative detection of DENV-2 mRNA after DENV-2 infection. With the increase titration of DENV-2, the expression of DENV-2 mRNA increased in the HUVEC group at 24 h, which was statistically significant (p < 0.05). After inhibition by bafilomycin A1, the expression of DENV-2 mRNA in infected group (1 × 102, 1 × 103 and 1 × 104 TCID50) increased and showed an upward trend, which was statistically significant (p < 0.05). Compared with the inhibition without bafilomycin A1, the expression of DENV-2 mRNA in HUVEC was decreased, which was statistically significant (p < 0.05).

Analysis of LC3-II expression in HUVECs infected with DENV-2 or treated with rapamycin (RAPA) or Bafilomycin A1.
Expression of autophagic marker protein LC3B detected by western blot in 24 h
As shown in Figure 2B–E, western blot was used to detect changes in the ratio of LC3II/I in 24 h after DENV-2 infection. Compared with the control group, the LC3BII/I ratio increased in the 1 × 102, 1 × 103, and 1 × 104 TCID50 DENV-2 infection group, which was statistically significant (p < 0.05). Compared with 1 × 103 TCID50 DENV-2 infection group, the grayscale of LC3B II and I bands decreased in 1 × 104 TCID50 DENV-2 infection group. After inhibition by bafilomycin A1, compared with the control group and the inhibition with bafilomycin A1 group, the ratio of LC3BII/I in the infected group (1 × 102, 1 × 103, 1 × 104 TCID50) increased.
Laser confocal scanning microscopy was used to detect DENV-2 infection of HUVEC autophagy marker protein LC3B, lysosomal marker protein LAMP1, DENV-2 NS1
As shown in Figure 3A, LC3B stained in green, LAMP1 stained in red, NS1 stained in orange, and DAPI stained cell nuclei in blue. HUVEC cytoplasm produced the fluorescence of LC3B and LAMP1; As shown in Figure 3B, the cytoplasm of DENV-2 infection group (1 × 104 TCID50) can observe the fluorescence of LC3B, LAMP1, and DENV-2 NS1, compared with the control group, the fluorescence of LC3B and LAMP1 increased significantly, the fluorescence of LC3B, LAMP1, and DENV-2 NS1 overlapped; As shown in Figure 3C, the fluorescence of LC3B and LAMP1 in the rapamycin-induced group were significantly enhanced compared with the normal control group.
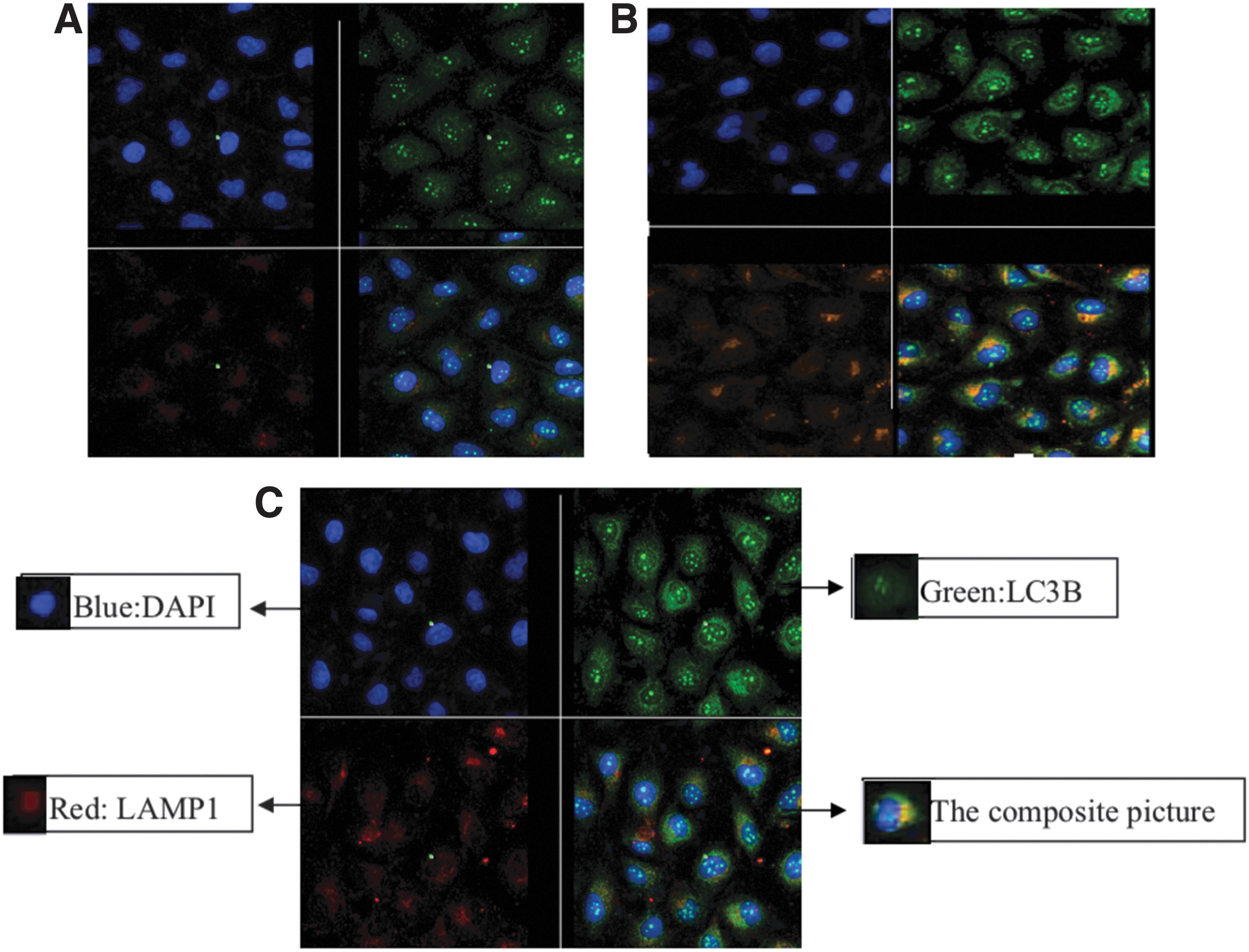
Autophagosomes in HUVECs infected with DENV-2 detected by laser confocal microscopy. HUVECs were infected by1 × 104 TCID50 DENV-2. LC3B stained in green, LAMP1 stained in red, NS1 stained in orange, and DAPI stained cell nuclei in blue. The cytoplasm of DENV-2 infection group (1 × 104 TCID50) can observe the fluorescence of LC3B, LAMP1 and DENV-2 NS1, compared with the control group, the fluorescence of LC3B and LAMP1 increased significantly, the fluorescence of LC3B, LAMP1, and DENV-2 NS1 overlapped.
Western blotting analysis of the change expression of LC3B, mTOR and p-Ser2448-mTOR of HUVEC in DENV-2 infection
The mechanism by which DENV-2 infection induces autophagy in HUVECs is not clear. The mTOR signaling pathway is the main pathway that regulates autophagy; therefore, we used western blotting to analyze whether DENV-2 infection inhibited mTOR activity. HUVECs were treated with rapamycin (RAPA) for 12 h, then infected with DENV-2, and proteins were harvested at 24 h; untreated HUVECs served as a negative control. As shown in Figure 4A and B, compared with the control group, the level of phosphorylated mTOR in the DENV-2-infected group or the RAPA-treated group decreased significantly (p < 0.05) (the total mTOR level did not change). The results confirmed that DENV-2 infection inhibited mTOR signaling; however, the level of LC3-II increased significantly (p < 0.05).

DENV-2 induces autophagy by inhibiting mTOR signaling pathway.
Western blotting analysis of the change expression of LC3B, p-Atg13, and p-ULK1 of HUVEC in DENV-2 infection
HUVECs were treated with rapamycin (RAPA) for 12 h, then infected with DENV-2, and proteins were harvested at 24 h; untreated HUVECs served as a negative control. As shown in Figure 5A and B, compared with the control group, the level of phosphorylated -Atg13 and p-ULK1 in the DENV-2-infected group or the rapamycin-treated group decreased significantly (p < 0.05); However, the level of LC3-II increased significantly (p < 0.05).
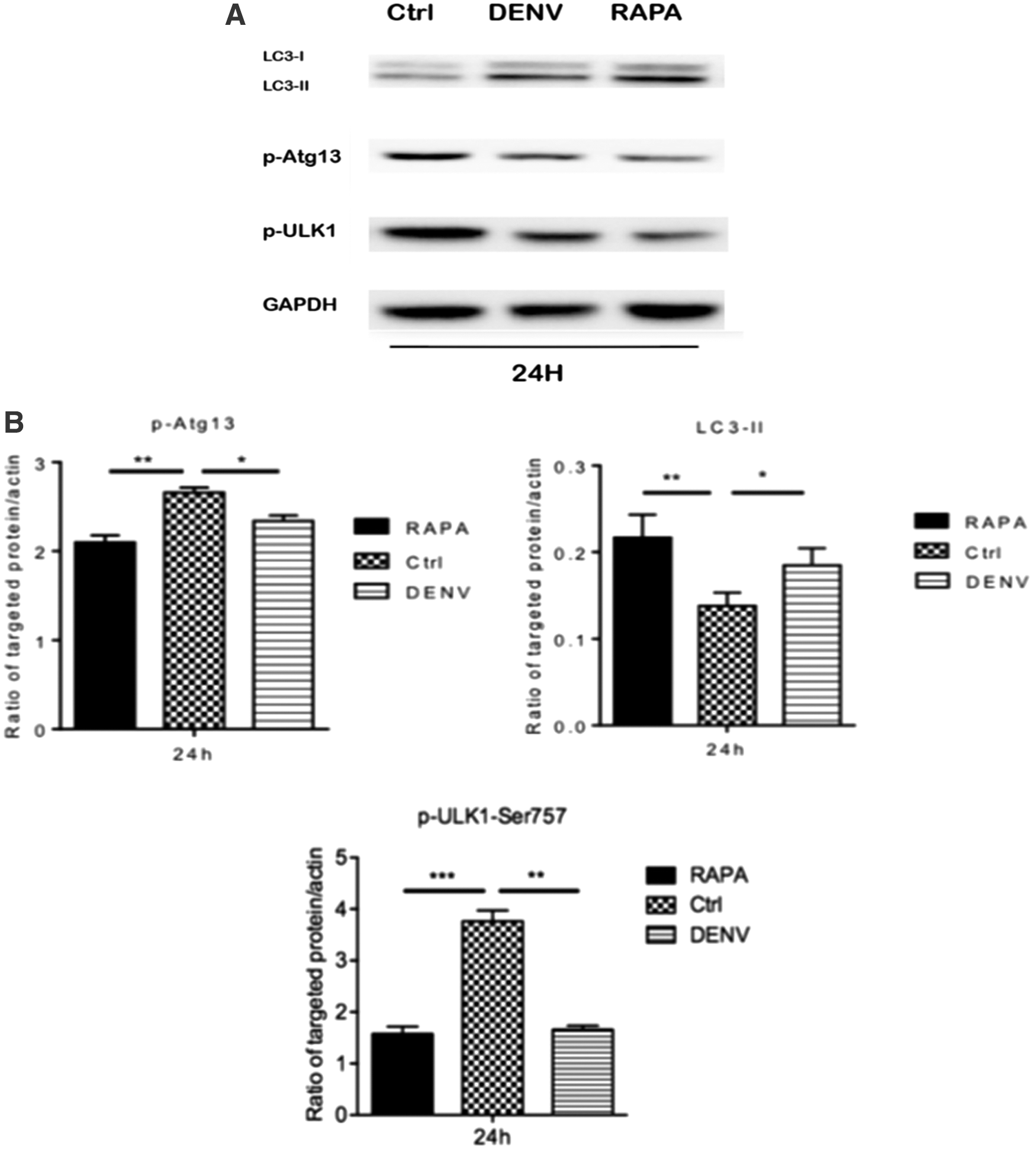
Analysis of expression of LC3-II, the phosphorylation levels of Atg13 (p-Atg13), and the phosphorylation levels of ULK1 (p-ULK1) in HUVECs infected with DENV-2 or treated with rapamycin (RAPA) for 24 h.
Discussion
DF is an acute infectious disease transmitted by Anopheles mosquitoes and caused by DENV (16,24). The clinical features are high fever; pain in muscles, joints, and bone marrow; skin rash; bleeding tendency; and lymphadenectasis. DHF and DSS are serious clinical symptoms and high mortality. Pathological changes of dengue hemorrhagic fever are systemic microvascular damage, resulting in plasma protein exudation and hemorrhage (9,28,31,34). Little is known about autophagy in human diseases. Autophagy may act as a compensatory mechanism to maintain homeostasis in the early stages of disease (10,15). Autophagy occurs in the late stages of disease progression and can induce sustained adaptive response or inadequate defense to block disease progression. Autophagy may play an important role in the pathogenesis of diseases, but its imbalance or overactivation directly affects cell death (30). In fact, many recent studies in cardiac injury or disease have shown that whether autophagy plays a protective or harmful role depends on the model system (36). Many studies have similarly argued that the significance of autophagy in these diseases is an open question.
Studies have shown that certain viruses can use autophagy to provide an advantage for their own replication and survival (11). Poliovirus and equine arteritis virus infection (positive strand RNA virus) can induce autophagy (8). SiRNA reduced the expression of Atg12 and LC3 (two key autophagy genes), which not only reduced the replication of poliovirus and rhinovirus outside the cell (12) but also reduced the viral load in the cell (27). Autophagy enhancement in autophagy is induced by NSP4 proteins. Parvovirus B19, single-stranded DNA virus, hepatitis A, B, and C virus induce autophagy as a means of prolonging the life of infected cells (29,37). Together, these studies suggest that viruses have evolved the ability to use cellular autophagy to promote viral replication. The extent to which viral replication depends on autophagy needs further investigation.
Whether DENV-2 can induce autophagy or the effect of autophagy on virus is the focus of this study, which provides scientific basis for the study of the mechanism of DENV-2 infection injury to vascular endothelial cells. In this study, human vascular endothelial cells (HUVEC) were used as a model of ATCC-ATCC Catalog No. CRL-1730TM. To identify HUVEC, the expression of factor VIII was detected by immunohistochemical method. The cell line was identified as HUVEC. To study the effect of DENV-2 infection of HUVEC on autophagy, the effect of DENV-2 infection in HUVEC on autophagy was studied by means of autophagy inhibitor. It has been proved that the degradation of autophagy protein was inhibited, the number of autophagy increased, and the number of autophagy lysosomes decreased after the occurrence of autophagy in tumor cells (18). It was proved that the fusion of autophagy and lysosome was blocked in the process of autophagy and lysosome fusion (14). The autophagy inhibitor used in this study was bavaromycin A1 (Bafilomycin A1).
There are two ubiquitin regulatory pathways in autophagy formation: the first is the ubiquitin pathway, in which autophagic gene 12 (Atg12) covalently binds to Atg7, Atg10, and Atg5 after activation, and then covalently binds with Atg16 to form Atg5-Atg12-Atg16 complex in the autophagic isolation membrane (7), and the second ubiquitin regulatory pathway is the ubiquitin regulatory pathway. The microtubule-associated carboxyl terminal amino acid protein light chain 3 (LC3, encoded by Atg8 homologous chromosomes) was exposed to a conserved glycine residue by cysteine protease Atg4 decomposition and was necessary for autophagy formation (8,19). The instantaneous covalent binding of lytic LC3 with Atg7, Atg3, and phosphatidylethanolamine resulted in the degradation of LC3BII, LC3BII when autophagy combined with lysosome (6). The two ubiquitin pathways regulate and influence each other in the process of autophagy. The increase of LC3 synthesis during autophagy makes it a marker of autophagy. The ratio of LC3BII/I is a relatively perfect index to detect autophagy in recent years (1). In this study, autophagy was detected by the ratio of LC3BII/I, a marker protein of autophagy.
Western blot was used to detect the change of LC3BII/I ratio in 24 h after infection with DENV-2. Compared with the control group, DENV-2 infection group (1 × 102, 1 × 103, and 1 × 104 TCID50), the band of LC3BII gray increased, LC3BII/I ratio increased, indicating that DENV-2 can induce autophagy, with the increase of DENV-2 titer, autophagy increased. Compared with 1 × 103 TCID50 DENV-2 infection group, the gray scale of LC3BII and I bands decreased in 1 × 104 TCID50 DENV-2 infection group.
After treated with rapamycin and bavaromycin A1, compared with the control, the gray scale of LC3BII and I band and the ratio of LC3BII/I increased in the DENV-2 infection (1 × 102, 1 × 103, and 1 × 104 TCID50), which indicated that DENV-2 could induce autophagy in HUVEC.
To see the occurrence of autophagy more intuitively, the phenomenon of autophagy was observed by laser confocal technique. Laser confocal scanning microscope can be used for immunofluorescence analysis of fixed cells to obtain specific antibody recognition target molecule expression, location, distribution changes, and other information. LC3B is a marker of autophagy in the process of autophagy formation. In the process of autophagy fusion with lysosome, lysosome was formed, and LAMP1 and LAMP2 (5), LAMP1 were obtained as lysosome marker proteins. The target molecules selected for laser confocal autophagy detection were autophagy-labeled protein LC3B (green fluorescent label) and lysosome-labeled protein LAMP1 (red fluorescent label). The selected marker protein of DENV was DENV-2 NS1 (orange fluorescent labeling).
Compared with the control group, the intensity of the fluorescence of autophagy-labeled protein LC3B and lysosome-labeled protein LAMP1 in the cytoplasm of HUVECs in the positive control group (rapamycin group) increased obviously, which indicated that the autophagy in HUVEC induced by rapamycin was enhanced. The cytoplasm of DENV-2 infection group (1 × 104 TCID50) can observe the fluorescence of LC3B, LAMP1, and DENV-2 NS1, compared with the control group, the fluorescence of LC3B and LAMP1 increased significantly, the fluorescence of LC3B, LAMP1, and DENV-2 NS1 overlapped.
Based on the question of whether DENV-2 can utilize HUVEC autophagy to promote DENV-2 replication, real-time fluorescent quantitative PCR was carried out to detect the effect of autophagy on DENV-2 replication at molecular level. The results showed that the DENV-2 mRNA content of HUVECs increased with the increase of DENV-2 infection concentration, and the DENV-2 mRNA content of HUVECs increased with the increase of infection concentration of DENV-2 after being inhibited by bavaromycin A1, compared with the inhibition of HUVECs not treated with bavaromycin A1; The decrease of DENV-2 mRNA content in HUVEC infected with the same concentration of DENV-2 indicated that HUVEC autophagy was enhanced, which was beneficial to the replication of DENV-2.
It is now generally accepted that mTOR is an important “inhibitor” of autophagy upstream (4), although the mechanism is not clear. The mechanisms of mTOR and autophagy signal pathway have little attention to dengue virus-2 (DENV-2) infection, especially the damage of HUVECs, and its regulation mechanism has not been clarified. However, there is evidence that mTOR is not the only regulator of autophagy upstream, adding ammonia from glutamine can induce autophagy activation. In mammals, ULK1 closely binds with Atg13 and FIP200 to form a complex. mTORCl is associated with nutritional dependence. ULK l/Atg13/FIP200 complex tightly binds to inhibit the occurrence of autophagy. mTORC l can phosphorylate ULK l and Atg13 and inhibit their function in autophagy.
In our study, we found that DENV-2 induces autophagy by inhibiting mTOR pathway, DENV-2 infection decreased the expression of p-Atgl3 and p-ULK1 and induced autophagy by inhibiting mTOR, so that the activity of mTORC l was not suppressed and the expression of p-Atgl3 and p-ULK1 was increased under the condition of drug stimulation in the future. It provides a basis for future research to protect endothelial cells by inhibiting autophagy.
Current studies have confirmed that mTOR and autophagy are involved in the occurrence and development of DENV disease, but the detailed mechanism of its regulation has not been explored in depth, although the organism can use cell autophagy to remove intracellular microorganisms. But disease-causing organisms have also evolved a sophisticated system to adapt to this cellular mechanism. For DENV-2, it is the use of cell autophagy for providing a theoretical basis for the injury mechanism of vascular endothelial cells infected by DENV-2.
Conclusion
We conclude that enhancement of autophagy in HUVEC is induced by DENV-2 infection. The mTOR signaling molecule plays an important role in DENV-2-mediated autophagy in HUVEC. The results provide a theoretical basis for the mTOR signal molecule to regulate the autophagy signaling pathway and regulate the process of viral infection.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (81560263; 81860289).