
Abstract
Follicular CD4+ T cells are the main HIV reservoirs due to, among other factors, the low frequency of CD8+ T cells in lymphoid follicles. Follicular CXCR5+ CD8+ T cells are associated with HIV control, but their differentiation conditions are yet undefined. In this study, we explored the in vitro effect of transforming growth factor (TGF)-β1, interleukin (IL)-12, and IL-23 on the induction of CXCR5, the follicle homing receptor, in human circulating CD8+ T cells from seronegative, and treated HIV-infected individuals. The combination of TGF-β1 plus IL-23 induced the highest expression of CXCR5 in purified CD8+ T cells. These CXCR5+ CD8+ T cells also expressed a transcriptional and phenotypic profile similar to that of follicular CD4+ T cells, such as the upregulation of BCL6, inducible costimulator and CD40L, and downregulation of PRDM1. These cells responded in vitro to CXCL13 and had low expression of CCR7. In addition, after polyclonal stimulation, they produced IL-21, interferon-γ, and de novo perforin. However, in comparison with seronegative individuals, CD8+ T cells from HIV-infected patients had a lower response to TGF-β1/IL-23, a defect that was restored with the blockade of the programmed cell death 1 inhibitory receptor. Thus, TGF-β1 plus IL-23 induce follicular-like CXCR5+ CD8+ T cells in seronegative individuals, but in HIV-infected patients there is a limited response which could impair the generation of this cell population.
Introduction
For lymphoid follicles homing, T cells have to increase the expression of the CXCR5 receptor, which responds to CXCL13 (40). Follicular CXCR5+ CD8+ T cells have been described (31) and characterized by the expression of the transcription factor B cell lymphoma 6 protein (BCL6), and low expression of the B-lymphocyte-induced maturation protein-1 (BLIMP-1) and CCR7 (44). However, the polarizing conditions to a follicular profile in human CD8+ T cells are undefined. Most of the available data come from human CD4+ T cells, and nonhuman primate CD8+ T cells, where a combination of transforming growth factor (TGF)-β1, interleukin (IL)-12, and IL-23 induced a follicular-like profile (42,52).
During HIV infection, there is an expansion of follicular CXCR5+ CD8+ T cells (22,46); remarkably, the magnitude of simian immunodeficiency virus (SIV)-specific CXCR5+ CD8+ T cells in lymph nodes, and human circulating CXCR5+ CD8+ T cells, are associated with limited viral replication (22,42). Due to their antiviral potential, this subset has attracted attention, as it could play an important role in the control of HIV replication and viral reservoirs in lymphoid follicles (27).
These findings provide the rationale for improving the ability of CD8+ T cells to migrate into lymphoid follicles through the induction of the expression of CXCR5, a strategy that could contribute to the elimination of HIV-infected follicular cells. Supporting this assumption, in SIV infection models, the transduction of CD8+ T cells with CXCR5 increased their migration into lymphoid follicles, in proximity to SIV-infected cells (3). In addition, SIV-specific chimeric antigen receptor-CD8+ T cells coexpressing CXCR5 migrated into follicles, exhibiting potent suppression of SIV replication in vitro (20).
Considering that TGF-β1, IL-12, and IL-23 have been proposed as differentiation factors for follicular CXCR5+ human CD4+ T cells (52) and nonhuman primate CD8+ T cells (42), but their effect is unknown on human CD8+ T cells, here we evaluated these cytokines as in vitro CXCR5-inducing factors in human CD8+ T cells from seronegative and HIV-infected individuals. The combination of TGF-β1 and IL-23 induced follicular-like CXCR5+ CD8+ T cells from seronegative individuals. Nonetheless, the programmed cell death 1 (PD-1) receptor limited the response of CD8+ T cells from HIV-infected patients.
Materials and Methods
Patients and samples
This study and written informed consent were approved by the Institutional Review Board of Universidad de Antioquia (certificates 15-08-634 and 11-08-352). All experiments followed the principles expressed in the Declaration of Helsinki. All the individuals signed the informed consent.
Two groups of individuals were included (Table 1): (1) HIV-infected patients who had received only one antiretroviral therapy (ART) combination for more than 1 year, with viral load <20 HIV RNA copies/mL and without previous therapeutic failure; (2) seronegative individuals, included based on a negative HIV test, as well as normal blood cell counts and the lack of clinically evident conditions, including HBV infection (such as jaundice, fever, or hepatomegaly). From each individual, 100 mL of venous blood was collected and anticoagulated with ethylenediaminetetraacetic acid (EDTA). The plasma was used for determining viral load with the clinical diagnostic test reverse transcriptase-polymerase chain reaction (RT-PCR) Ampliprep–Cobas (Roche), with a detection limit of 20 copies/mL.
Characteristics of the Individuals Included
All HIV-infected patients had undetectable viral load.
ART, antiretroviral therapy; N/A, does not apply.
Induction of follicular-like CXCR5+ CD8+ T cells in vitro
In this study, we aimed to evaluate the in vitro effect of TGF-β1, IL-12, and IL-23 in the setting of T cell receptor (TCR)-mediated stimulation to partially mimic the CD8+ T cell priming mediated by an antigen-presenting cell, as previously reported for the differentiation of CD8+ T cell subsets (24,42). After peripheral blood mononuclear cells (PBMC) isolation using the Ficoll density gradient, CD8+ T cells were purified through negative magnetic cell sorting, using the CD8+ T cell Isolation Kit (Miltenyi Biotec); a purity >90% was obtained in all the experiments.
After isolation, CD8+ T cells were resuspended in RPMI-1640 supplemented with 10% fetal bovine serum, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 2 mM L-glutamine and stained with CellTrace CFSE (all from Thermo Fisher). Then, 2 × 106 cells/mL were separately stimulated in 24-well flat bottom plates (Sigma-Aldrich) with mouse anti-human CD3 (plate-bound for 2 h at 37°C) and soluble mouse anti-human CD28 (both at 1 μg/mL; clones OKT3 and CD28.2, respectively; Thermo Fisher), in the presence or absence of human recombinant TGF-β1, IL-23, and/or IL-12 (Thermo Fisher), and incubated for 5 days at 37°C in 5% CO2. Finally, the cells were collected for the detection of CXCR5-expressing cells by flow cytometry, as described below.
After culture, the cell viability was >85%, as evaluated by the Fixable Viability Dye eFluor 506 (Thermo Fisher). In some experiments, purified CD8+ T cells from HIV-infected patients were mixed 1:1 with autologous total PBMC and cultured for 5 days with hrTGF-β1 plus IL-23, in presence or absence of anti-human PD-1 (clone J116; Thermo Fisher) or hrPD-L1 hIgG1-Fc Tag (Sino Biological).
In this study, we report the relative frequency of CXCR5+ CD8+ T cells (% of the population) with each evaluated condition, as well as the % fold change in the frequency of this subset with anti-CD3/CD28+cytokine treatments, relative to the treatment with anti-CD3/CD28 alone, as follows: % fold change in CXCR5+ CD8+ T cells = ([anti-CD3/CD28/cytokines treatment − anti-CD3/CD28 alone condition]/[anti-CD3/CD28 alone condition]) × 100.
Detection of CXCR5+ CD8+ T cells
CXCR5+ CD8+ T cells in whole blood and within total purified CD8+ T cells were detected by flow cytometry. The whole blood staining protocol was previously described (45). For purified CD8+ T cells, after cell culture, 5 × 105–1 × 106 cells were incubated for 30 min at room temperature with optimized doses of the following anti-human antibodies from Thermo Fisher: mouse anti-CD8-Alexa Fluor 700 (clone OKT8), rat anti-CCR7-PE (clone 3D12); and from BD: rat anti-CXCR5-PerCP Cy 5.5 (clone RF8B2); mouse anti-PD-1- BV510 (clone EH12.1); mouse anti-CD40L-APC-Cy7 (clone TRAP1); mouse anti-inducible T cell costimulator (ICOS)-Alexa Fluor 647 (clone DX29).
Moreover, in a fraction of the individuals, the APC- and PE-labeled anti-TGF-β1 receptor II (TGF-βRII) and anti-IL-23 receptor (IL-23R) antibodies, respectively, were used (both from R&D). Next, the cells were washed with 1 mL of 1 × phosphate-buffered saline (PBS) and resuspended in 1% paraformaldehyde. The cells were acquired on an LSR Fortessa cytometer (BD), using the FACS Diva software v.6.0, within an hour of completing the staining. At least 100,000 CD8+ singlet events (FSC-A vs. FSC-H) were acquired. The data were analyzed with the FlowJo Software version 10.4 (Tree Star, Inc.). Fluorescence minus one control was included to define positive thresholds.
Functionality of follicular-like CXCR5+ CD8+ T cells
After 5 days of culture, under the follicular-inducing conditions, purified CD8+ T cells were restimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin (at 50 and 500 ng/mL, respectively; both from Sigma-Aldrich), and incubated for 6 h at 37°C in 5% CO2, all in the presence of 5 μg/mL of brefeldin A and monensin (both from Thermo Fisher), as well as an optimized dose of the APC-labeled mouse anti-human CD107a antibody (clone H4A3; BD).
After incubation, the cells were harvested and washed with 1 mL of 1 × PBS. Next, lineage antibody cocktail for cell surface staining was added and incubated for 30 min at 4°C, light-protected, followed by a wash, and cell fixation and permeabilization with the Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher). Finally, the intracellular expression of IL-21, IFN-γ, and perforin was evaluated by flow cytometry, using the mouse anti-human IL-21-BV421 (clone 3A3-N2.1; BD), mouse anti-human IFN-γ-PE-Cy7 (clone 4S.B3; Thermo Fisher), and mouse anti-human perforin-PE (clone B-D48; BioLegend), respectively. Fluorescence minus one control was also included.
In addition, purified CD8+ T cells were stimulated without brefeldin A and monensin, and after the culture, supernatants were collected for the detection of soluble molecules using the human granzyme B, IFN-γ, and CCL5 flex set kits (BD), with detection limits (in pg/mL) of 4, 1.8, and 0.002, respectively.
In vitro transwell migration assay
Purified CD8+ T cells, treated as described, were washed thoroughly and 2 × 105 cells were resuspended in 200 μL of complete medium, and placed in the upper chamber of a 24-well plate, with a 3 μm polyester transwell membrane (Sigma-Aldrich). To the lower chamber, 2.5 μg/mL of hrCXCL13 (R&D) in a total volume of 600 μL/well was added. No chemokine was added to control wells. After incubation for 4 h at 37°C in 5% CO2, cells were collected from the lower chamber, counted, stained with lineage antibodies and acquired in a flow cytometer. Specific cell migration was determined by subtracting the percentage of cells (CXCR5+ or CXCR5−) that migrated to media alone from the number of cells that migrated to the chemokine.
Gene expression analysis
The expression of BCL6, PRDM1, TCF7, ID2, ID3, IL21, CXCR5, and GZMB in purified CD8+ T cells after in vitro treatments was analyzed by semiquantitative RT-PCR, as previously described (13). Oligonucleotides sequences are shown in Supplementary Table S1. The gene relative expression induced by the anti-CD3/CD28+cytokine treatment was calculated with the ΔΔCt method, using for normalization the β-actin gene and cells treated with anti-CD3/CD28 alone.
Statistical analysis
Data are presented as medians and ranges. The Mann–Whitney and Wilcoxon tests were used for comparison of two independent and paired data, respectively. The Friedman and Dunn's post hoc tests were used for comparison of three or more groups. The Spearman test was used for correlation analyses. In all cases, a p value <0.05 was considered significant.
Results
TGF-β1 plus IL-23 induce the expression of CXCR5 in human CD8+ T cells
First, we evaluated the phenotypic profile of CXCR5-expressing CD8+ T cells from seronegative individuals. In addition to CXCR5, we evaluated the expression of the chemokine receptor CCR7, which induces the migration of T cells to extrafollicular zones (15), ICOS, and CD40L, involved in the interaction between follicular T cells and B cells (19,55), and PD-1, which is highly expressed by follicular T cells (21), and involved in the regulation of immune responses (63). CXCR5+ CD8+ T cells exhibited a low expression of CCR7, ICOS, and CD40L, but more than half of them expressed PD-1 (Fig. 1A).

TGF-β1 plus IL-23 induce the expression of CXCR5 in human CD8+ T cells.
Next, we evaluated the in vitro effect of TGF-β1, IL-12, and IL-23 on the induction of CXCR5 expression in human CD8+ T cells. Purified CD8+ T cells from seronegative individuals were cultured with anti-CD3/CD28 antibodies alone (as control) or plus the cytokines, independently or in combination. Of note, since the anti-CD3/CD28 stimulation induces differential regulation of the evaluated parameters, our analyses were focused on the comparison between the anti-CD3/CD28+cytokine versus the anti-CD3/CD28 alone conditions.
A low but detectable frequency of CXCR5+ cells was observed among purified CD8+ T cells in the untreated condition (Fig. 1B), consistent with a low frequency of this population in circulation (45,47). The anti-CD3/CD28 alone treatment increased, although nonsignificantly, the expression of CXCR5 in CD8+ T cells (Fig. 1B). When each cytokine was evaluated separately, only TGF-β1 induced the upregulation of CXCR5, whereas IL-12 and IL-23 downregulated its expression, independently or in combination (Fig. 1C). Interestingly, the addition of IL-12 or IL-23 potentiated the effect of TGF-β1, being higher with the combination of TGF-β1 and IL-23 (Fig. 1B, C). However, the combination of the three cytokines did not yield an increase of the frequency of CXCR5+ CD8+ T cells (Fig. 1C).
Thus, the combination of TGF-β1 and IL-23 induced the highest upregulation of CXCR5 in human CD8+ T cells (Fig. 1B, C), an effect that was confirmed by quantitative PCR (Fig. 1D) and was dose-dependent (Fig. 1E). Of note, we did not observe statistically significant differences in the % of CXCR5+ CD8+ T cells that were CFSElo between cells treated with anti-CD3/CD28 alone versus anti-CD3/CD28+TGF-β1/IL-23 (p = 0.8, Mann–Whitney test), suggesting that the increase in the % of CXCR5+ CD8+ T cells with TGF-β1/IL-23 is not due to preferential proliferation of this population with the cytokine treatment. The concentrations of 15 ng/mL of TGF-β1 and 75 ng/mL of IL-23 were chosen for the subsequent experiments.
TGF-β1 plus IL-23 induce a follicular-like profile in human CXCR5+ CD8+ T cells
In addition to CXCR5, the expression of CCR7 also influences the migration of T cells into lymphoid follicles (21). Circulating CXCR5+ CD8+ T cells express CD62L to migrate into secondary lymphoid organs (45), and have a low expression of CCR7 (Fig. 1A) (45), consistent with a potential follicle migration ability. Accordingly, untreated, anti-CD3/CD28 alone-treated and anti-CD3/CD28 alone+TGF-β1/IL-23-treated CXCR5+ CD8+ T cells exhibited low expression of CCR7 (Fig. 2A).
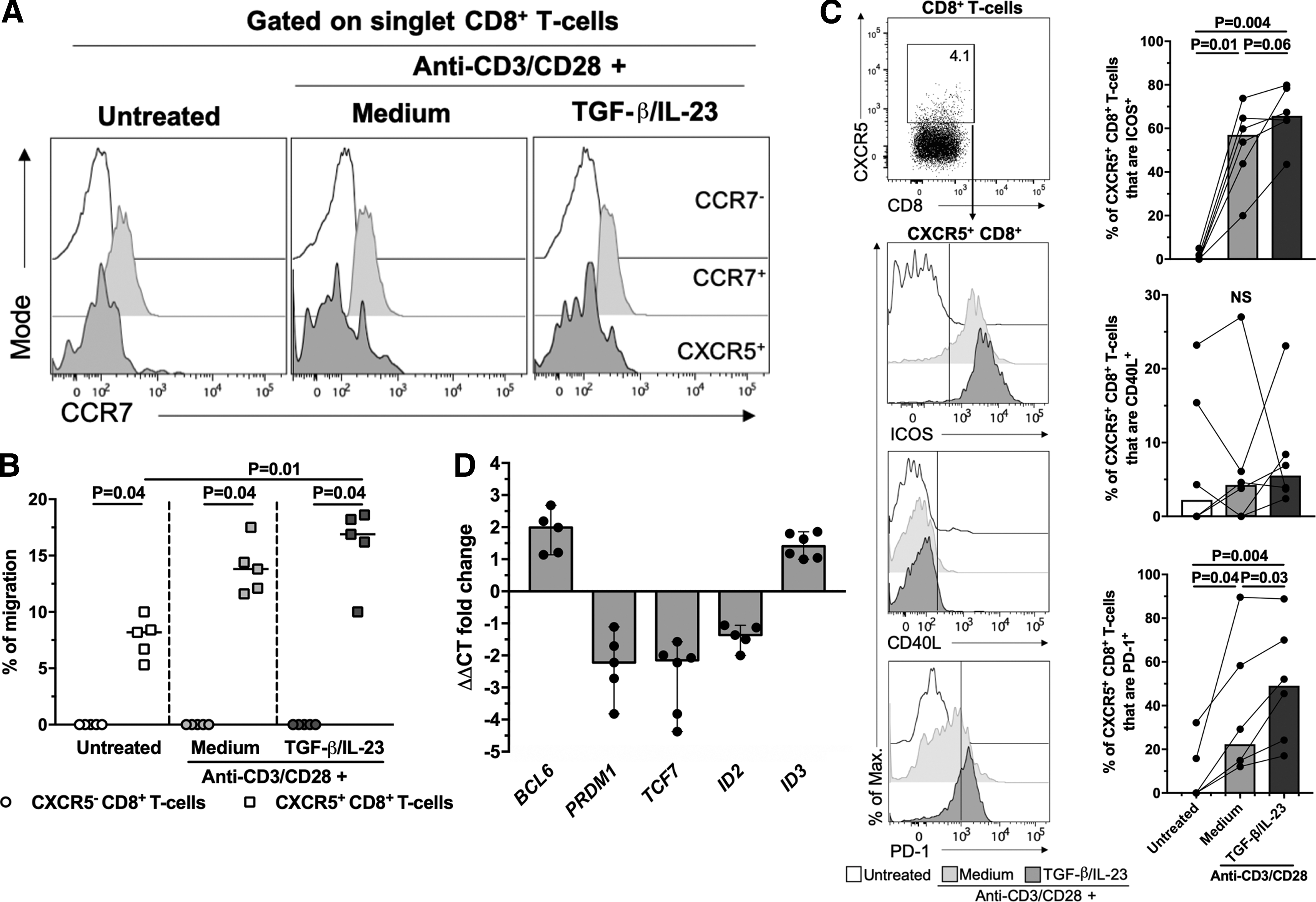
TGF-β1 plus IL-23 induce follicular-like CXCR5+ CD8+ T cells.
Moreover, CXCR5+ CD8+ T cells, but not CXCR5− CD8+ T cells, migrated in response to CXCL13 in a transwell assay, and the percentage of migration was higher in cells treated with TGF-β1/IL-23 compared with untreated cells, but not with the anti-CD3/CD28 alone (Fig. 2B). The latter result was related with a higher density of expression of the CXCR5 molecule with the cytokine treatment, as we observed a positive correlation between CXCR5 median fluorescence intensities (MeFI) in total CD8+ T cells and the percentage of migration of CXCR5+ CD8+ T cells (ρ = 0.79, p = 0.01, Spearman test).
To explore the effect of TGF-β1/IL-23 on the expression of other follicular T cell markers, we evaluated the expression of ICOS, CD40L, and PD-1 in CD8+ T cells after in vitro treatments. Consistent with previous reports (1,6,25), the stimulation with anti-CD3/CD28 antibodies increased the frequency of total CD8+ T cells expressing ICOS, CD40L, and PD-1 and/or their respective MeFI (Supplementary Fig. S1A). Similar results were observed for CXCR5+ CD8+ T cells, except for the case of CD40L (Fig. 2C and Supplementary Fig. S1B). Interestingly, compared with cells treated with anti-CD3/CD28 antibodies alone, the addition of TGF-β1/IL-23 increased the frequency and MeFI of CXCR5+ CD8+ T cells that are ICOS+ and PD-1+ (Fig. 2C and Supplementary Fig. S1B), and the MeFI of ICOS, CD40L, and PD-1 in total CD8+ T cells (Supplementary Fig. S1A).
Ultimately, we evaluated the expression of BCL6 (which codifies for BCL6), PRDM1 (which codifies for BLIMP-1), TCF7 (which codifies for T cell Factor 1 [TCF-1]), ID2, and ID3 (which codify for inhibitor of differentiation (Id)2 and Id3, respectively), which are differentially expressed in follicular CXCR5+ CD8+ T cells (26,31). TGF-β1/IL-23 upregulated the expression of BCL6 and ID3, whereas downregulated the expression of PRDM1, TCF7, and ID2 (Fig. 2D). Altogether, these results support that TGF-β1/IL-23 induces a follicular-like profile of human CD8+ T cells, which exhibit a potential follicle homing.
TGF-β1/IL-23-treated CXCR5+ CD8+ T cells exhibit lytic and nonlytic effector mechanisms
To evaluate effector mechanisms of CXCR5+ CD8+ T cells, after treatment with TGF-β1/IL-23, purified CD8+ T cells were restimulated with PMA-ionomycin and the expression of IL-21, granzyme B, perforin, CD107a, and IFN-γ was evaluated. There was a higher frequency of IL-21+ CXCR5+ CD8+ T cells after treatment with TGF-β1/IL-23 in comparison with the anti-CD3/CD28 alone condition or the untreated cells (Fig. 3A). In addition, there was a comparable de novo production of perforin and granzyme B (perforin+ or granzyme B+ CD107a+ cells), as well as IFN-γ (Fig. 3B and data not shown) by CXCR5+ CD8+ T cells between the conditions evaluated. We did not observe IL-21+ IFN-γ + cells (data not shown).
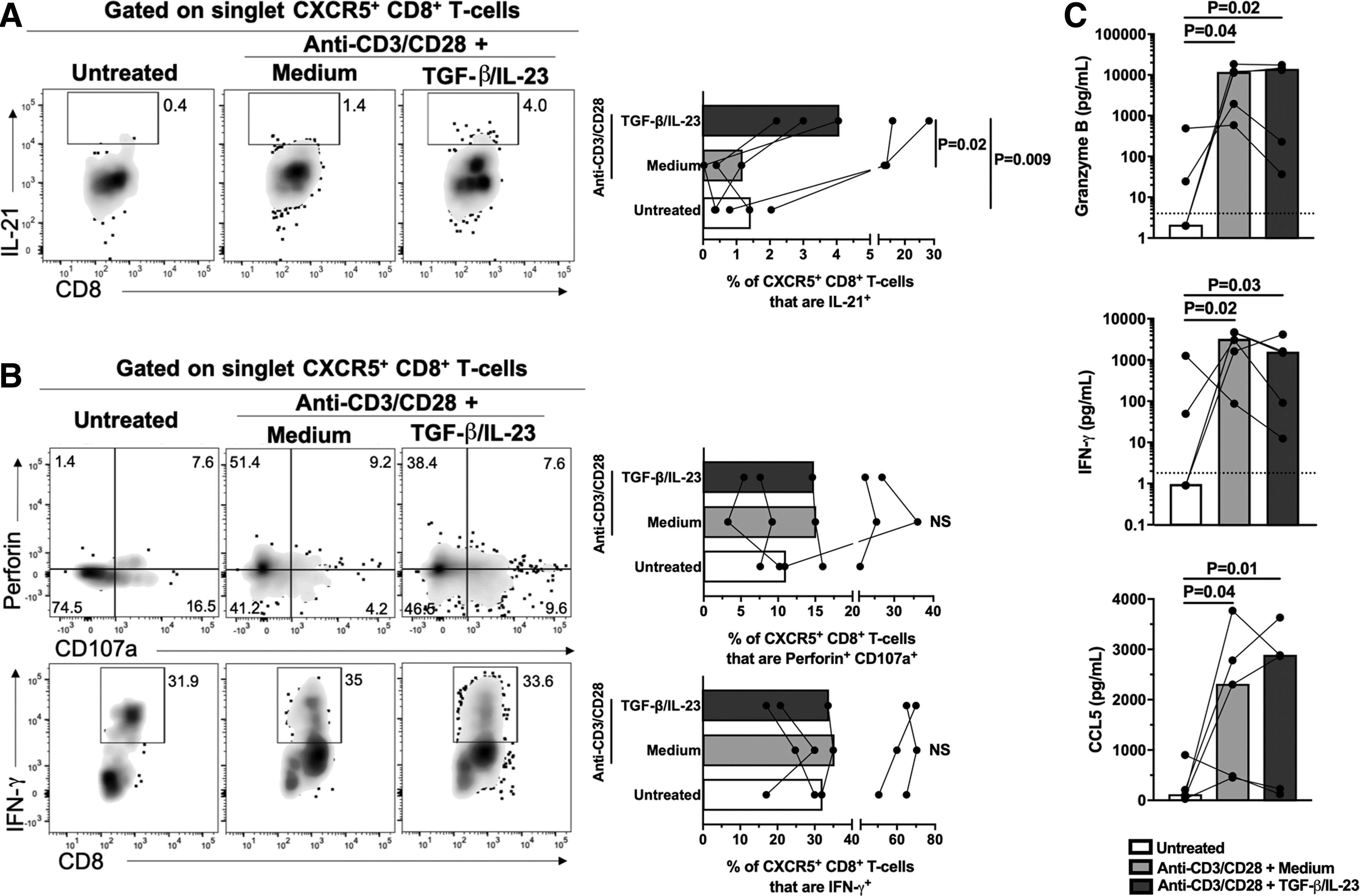
Functional capacity of follicular-like CXCR5+ CD8+ T cells.
Moreover, CD8+ T cells treated with TGF-β1/IL-23 or with anti-CD3/CD28 alone had a comparable secretion of granzyme B, IFN-γ, and CCL5, evaluated in culture supernatant (Fig. 3C). Finally, CD8+ T cells upregulated IL21 and GZMB (which codify for IL-21 and granzyme B, respectively) after treatment with TGF-β1/IL-23 (Supplementary Fig. S1C). Together, these results indicate that TGF-β1/IL-23-treated CXCR5+ CD8+ T cells exhibit lytic and nonlytic effector functions.
Low response of CD8+ T cells to TGF-β1/IL-23 during HIV infection
After establishing the in vitro effect of TGF-β1/IL-23 on CD8+ T cells, we evaluated the response of CD8+ T cells from HIV-infected patients to TGF-β1/IL-23. In basal conditions, the frequency of CXCR5+ CD8+ T cells was comparable between seronegative and HIV-infected individuals (Fig. 4A), consistent with a therapy-induced reconstitution of this subset (45). However, HIV-infected patients exhibited a lower upregulation of CXCR5 with TGF-β1/IL-23 (Fig. 4A, B).

Lower response of CD8+ T cells from HIV-infected patients to TGF-β1/IL-23.
CD8+ T cells treated with TGF-β1/IL-23 from seronegative and HIV-infected individuals had a comparable upregulation of BCL6 and downregulation of PRDM1, but HIV-infected patients had a significantly higher upregulation of ID3 (Supplementary Fig. S2A), without differences for TCF7 and ID2 (data not shown). Moreover, TGF-β1/IL-23-treated CD8+ T cells from HIV-infected patients had a negative regulation of IL21 and GZMB (Supplementary Fig. S2B). Moreover, TGF-β1/IL-23-treated CD8+ T cells from HIV-infected patients had lower supernatant levels of granzyme B, IFN-γ, and CCL5 in comparison with cells from seronegative individuals (Supplementary Fig. S2C). Thus, CD8+ T cells from HIV-infected patients have a lower response to TGF-β1/IL-23, in addition to a functional defect.
To explore the possible mechanisms responsible for the functional defect in CD8+ T cells from HIV-infected patients, we explored the expression of the TGF-βRII and IL-23R in CD8+ T cells from seronegative and HIV-infected individuals. Both receptors form heterodimers with the TGF-βRI (in the case of TGF-βRII) and IL-12Rβ1 (in the case of IL-23R) and mediate the response to TGF-β1 and IL-23, respectively. CD8+ T cells from seronegative and HIV-infected individuals exhibited similar levels of these receptors (Supplementary Fig. S2D), both in basal conditions and after stimulation with anti-CD3/CD28 and TGF-β1/IL-23 (data not shown), indicating that the low response of CD8+ T cells from HIV-infected patients to TGF-β1/IL-23 is not due to a decreased expression of their receptors.
Next, we pointed to PD-1, which impairs the response to T cell differentiation factors after binding to its ligands PD-L1 and PD-L2 (33) and is highly expressed by CD8+ T cells from HIV-infected patients (57). Indeed, in basal conditions, total CD8+ T cells from HIV-infected patients exhibited higher MeFI of PD-1 than cells from seronegative controls (Supplementary Fig. S2E). Treatment with TGF-β/IL-23 also increased the levels of PD-1 in CD8+ T cells from HIV-infected patients, but they were similar to those seen in seronegative individuals (Supplementary Fig. S2E).
We cultured purified CD8+ T cells from HIV-infected patients with TGF-β/IL-23, in the presence or absence of an anti-PD-1 monoclonal antibody, as well as hrPD-L1 to increase the ligand-receptor interaction. The coadministration of the anti-PD-1 antibody with the TGF-β1/IL-23 treatment promoted the upregulation of CXCR5 in CD8+ T cells from HIV-infected patients in a dose-dependent manner (Fig. 4C). Conversely, the treatment with the hrPD-L1 protein negatively affected the expression of CXCR5 (Fig. 4D). These results were not evidenced in cells purified from seronegative individuals (Fig. 4C, D).
Remarkably, the addition of anti-PD-1 to the TGF-β1/IL-23 treatment increased the % fold change in CXCR5+ CD8+ T cells from HIV-infected patients to similar levels to those in seronegative individuals (Fig. 4E), whereas promoted the in vitro migration of CXCR5+ CD8+ T cells (Fig. 4F). These results are consistent with a negative modulation of PD-1 on the response of CD8+ T cells to the TGF-β1/IL-23 treatment in HIV-infected patients.
Discussion
Total and HIV/SIV-specific CD8+ T cells are largely excluded from lymphoid follicles, where viral replication and reservoir seeding are increased, whereas they are mainly localized in extrafollicular zones (8,9,23). Indeed, depletion of CD8+ T cells dramatically increases viral replication in extrafollicular zones, remaining high in follicles (32). Therefore, important efforts are being made to promote the migration of CD8+ T cells to follicles, for eliminating HIV/SIV-infected cells.
Strategies to increase the CD8+ T cells expression of CXCR5 include genetic engineering (3,20) and IL-15 agonists (59), whereas the cytolytic response of CXCR5-expressing CD8+ T cells can be redirected via bispecific antibodies (14,46). Developing HIV vaccines are also being focused on inducing the response of antigen-specific CXCR5+ CD8+ T cells for enhancing viral control (58), highlighting the importance of boosting the CXCR5+ CD8+ T cell response as a strategy to improve HIV control.
Another strategy to promote the human CXCR5+ CD8+ T cells response is the use of cytokines such as TGF-β1/IL-23 (42,52). Accordingly, our results indicate that these cytokines induce follicular-like CXCR5+ CD8+ T cells, with capacity of migration in response to CXCL13, and with lytic and nonlytic effector mechanisms.
Of note, although naive cells are the most probable CD8+ T cell subset responding to the in vitro polarizing factors (10), effector and memory cells could eventually respond to TGF-β1/IL-23, as genes such as BCL6 are in a poised state for potential transcriptional activation in these populations (53). Therefore, the interaction between polarizing cytokines and their respective signaling cascades could induce different profiles in human CD8+ T cells (53). Nonetheless, it remains to be defined the response of single CD8+ T cell subsets, as well as cells confined to lymphoid tissues, since their differentiation profile could influence the response to polarizing conditions.
Moreover, it is important to note that, compared with antigen-specific stimulation (such as viral proteins), the anti-CD3/CD28 stimulation induces a more potent activation of CD8+ T cells, which could influence the polarization process of these cells, as reported for type 1 and 2 CD8+ T cells (28). Similarly, the type of antigen, its concentration, and the level of CD8+ T cell activation could affect the polarization of follicular-like CXCR5+ CD8+ T cells in vivo (5,29).
CD4+ T cell studies have demonstrated that the activation of the signal transducer and activator of transcription (STAT) 3 and 4 by IL-23 would be partially responsible for the transcriptional signature of follicular T cells (52). Although IL-12 can also activate the STAT3 and 4 pathways and sustain the expression of CXCR5 in activated CD4+ T cells (36), this would not be the case for circulating CD8+ T cells, as well as human tonsil CD8+ T cells (48).
Importantly, TGF-β1 target genes are largely unknown; however, some genes that regulate T cell differentiation, such as those codifying for the transcription factors T-bet and GATA-binding protein 3 (GATA3), are suppressed by TGF-β1 (17,18). On the contrary, TGF-β1 induces the expression of the transcription factors forkhead box p3 (Foxp3) and RAR-related orphan receptor gamma (RORγt) (12,37).
Interestingly, TGF-β1 increases Foxp3 expression by promoting the binding of the transcription factor E2A to the Foxp3 promoter (38). Taking into account that E2A binds to the CXCR5, BCL6, ID2, and ID3 loci, and is critical for follicular CXCR5+ CD8+ T cells differentiation (31), E2A activation could be a mechanism by which TGF-β1 promotes a follicular profile in human CD8+ T cells. Nonetheless, TGF-β1 noncanonical pathways and cross-talking with other signaling cascades could also be involved in the differentiation of follicular T cells (2).
Importantly, although TGF-β1/IL-23 treatment induced the expression of BCL6 and ID3 and downregulation of PRDM1 and ID2, a transcriptional profile characteristic of follicular T cells, we also observed downregulation of TCF7, which codifies for the TCF-1. This transcription factor is critical for follicular CXCR5+ T cell differentiation, as it promotes the expression of BCL6 and repression of PRDM1 (26,62). This result may be related with the suppression of TCF7 via STAT4 signaling (elicited by IL-12 and IL-23) in CD8+ T cells (11), that can be facilitated by the presence of an important proportion of effector cells among total CD8+ T cells, with low expression of TCF7 and a possibly more silent transcriptional state (26,30).
Similar to human CD4+ T cells (35,52), TGF-β1/IL-23 promoted the production of IL-21 by CXCR5+ CD8+ T cells. In addition to promoting B and follicular CD4+ T cell responses (54), IL-21 also increases the survival and function of CD8+ T cells (43,60). Furthermore, follicular-like CXCR5+ CD8+ T cells maintained their cytotoxic potential and production of IFN-γ after polyclonal stimulation. These findings contrast with the fact that TGF-β1 represses the cytotoxic program and IFN-γ production by CD8+ T cells (34,56), as well as BLIMP-1, which regulates this effector function (51). This issue could be explained by the significant pool of effector CD8+ T cells in circulation that apparently maintain their cytolytic program despite the TGF-β1/IL-23 treatment. These effector mechanisms are important for the antiviral CXCR5+ CD8+ T cell response and therapeutic strategies, since a lower cytotoxic potential is attributed to the follicle-confined CD8+ T cell subset (44).
Remarkably, according to its negative effect in the activation of T cells, and increased expression in CD8+ T cells from HIV-infected patients (57), PD-1 decreased the response of CD8+ T cells to TGF-β1/IL-23, most likely due to the inhibition of the TCR signaling (49), an effect that was restored with receptor blockade.
In this study, we showed that PD-L1-PD-1 interaction is responsible for the limited response of CD8+ T cells to the polarizing factors; however, it remains to be defined if this is similar for PD-L2-PD-1, or there is an amplification of the inhibitory effect when both ligands are present. In addition to this mechanism, the decrease in naive CD8+ T cells during HIV infection could limit the response to TGF-β1/IL-23 (50). The negative role of PD-1 in CD8+ T cells from HIV-infected patients was additionally supported by the lower expression of effector molecules.
Importantly, CXCR5+ CD8+ T cells are the main subset responding to PD-1 blockade during chronic infection in mice (26), and the in vivo blockade of PD-1 in SIV-infected macaques increases the magnitude of SIV-specific CXCR5+ CD8+ T cells in lymph nodes (41). In addition, PD-1 blockade could reactivate latent viral reservoirs, allowing recognition and elimination of these infected cells (16,61). Thus, the combination of TGF-β1/IL-23 treatment plus PD-1 blockade could be a potential strategy for the stimulation of CXCR5+ CD8+ T cell responses that could be used, for instance, in adoptive cell transfer therapy.
Finally, it is important to note that CXCL13 plasma levels are increased in untreated HIV-infected patients, and are associated with immune activation and disease progression, whereas the ART incompletely restores the levels of this chemokine (39). In this context, despite high CXCL13 levels and increased follicle activity, the impairment in the response of CD8+ T cells to TGF-β1/IL-23 may limit the coordinated migration of this population to follicles and an efficient antiviral function.
Since we used cells derived from HIV-infected patients under suppressive ART, it remains to be defined the response of CD8+ T cells from other groups of patients, such as progressor/noncontroller and nonprogressor/controller individuals. Nonetheless, based on a previous report where a higher frequency of circulating CXCR5+ CD8+ T cells was found in HIV controllers, compared with noncontroller patients (45), and considering the lower expression of PD-1 (7) and a less differentiated phenotype in CD8+ T cells in these individuals (4), we hypothesize that HIV controllers have a higher response to TGF-β1/IL-23 than HIV noncontrollers, and may represent a mechanism associated with their condition.
Footnotes
Acknowledgments
We thank Gustavo Castro, Kevin Leon, Jorge Lujan, and Thera Clinic for their help in patient recruitment. This study was supported by Universidad de Antioquia (UdeA), COLCIENCIAS (Code: 111577757051) and Corporación Universitaria Remington–Uniremington (Code: 4000000121-17).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.