
Abstract
Autophagy is a finely tuned process in the regulation of innate immunity to avoid excessive inflammatory responses and inflammasome signaling. In contrast, the results of recent studies have shown that autophagy may disease-dependently contribute to the pathogenesis of liver diseases, such as fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) during hepatitis B virus (HBV) infection. HBV has learned to subvert the cell's autophagic machinery to promote its replication. Given the great impact of the autophagy mechanism on the HBV infection and HCC, recognizing these factors may be offered new hope for human intervention and treatment of chronic HBV. This review focuses on recent findings viewing the dual role of autophagy plays in the pathogenesis of HBV infected hepatocytes.
Introduction
The hepatitis B virus (HBV) is an enveloped virus with partially double-stranded circular DNA belonging to the Hepadnaviridae family (12). Blumberg discovered the HBV in 1967 who won the Nobel Prize in medicine for his discovery in 1976 (10,11). Dane et al. (23) characterized the whole viral particle using electron microscopy in the 1970s. In 1980, the first commercially available HBV vaccine was approved by the FDA upon which the genome of HBV was sequenced (115). Beasley et al. (9) reported a strong association between HBV and hepatocellular carcinoma (HCC) in their breakthrough study of 22,707 men in Taiwan. Chronic hepatitis B is a major important leading cause of HCC (8).
HBV genome harbors four overlapping open reading frames and encodes seven viral proteins, including viral polymerase, core (HbcAg) and hepatitis B envelope antigen (HBeAg), regulatory X protein (HBx), and large (LHBs), medium (MHBs), and small (SHBs) variants of the surface antigens (HBsAg) (125). HBV is classified into 10 genotypes (A–J) based on the criteria of >8% genetic variances in the whole genome sequence and >4% in the S gene. Among the proteins of the virus, HBx as a multifunctional protein has a significant role in the regulation of viral replication and various cellular functions (85). Autophagy has been revealed to play an important role in HBV multiplication and pathogenesis (110).
Autophagy as an innate intracellular immunity has a pivotal role in pathogen clearance (28). HBV is one of the viruses that can increase its replication by subverting this mechanism. This interference increases the form of autophagosome (66). In addition, autophagy has a strong influence on carcinogenesis and tumor progression by altering the risk factors of liver cancer. Despite the growing body of researches, the HBV interactions with the host cells are not clearly understood (35). The lack of a permissive and efficient in vitro and animal infectivity model has been a major obstacle to study of HBV in a natural infection system (40). In this study, we discuss the relationship between HBV and autophagy mechanism, also pointed out various factors which are involved in this relationship.
Autophagy: Process and Function
Autophagy is a protective mechanism through which misfolded proteins, damaged organelles, and pathogens are engulfed in double layer name as autophagosome and transferred to the lysosome to degrade its components. Christian de Duve coined the term “autophagy” in 1963, at the Ciba Foundation symposium on lysosomes to describe the presence of single-membrane or double-membrane vesicles that enclose parts of the cytoplasm and organelles (25). The 2016 Nobel Prize in physiology or medicine was awarded to Yoshinori Ohsumi due to his discoveries of mechanisms for autophagy (44).
This mechanism is regulated by approximately more than 40 autophagy-related genes (ATGs) and according to the type of delivery mechanism to lysosomes to degrade are divided into three types: microautophagy, chaperone-mediated autophagy, and macroautophagy (3,7). In general, autophagy is activated in an undesirable condition or particular stress stimuli such as starvation, hypoxia, endoplasmic reticulum (ER) stress, increased reactive oxygen species (ROS) levels, infection, and chemotherapy (64). During this process, a double membrane isolated from the cytoplasm enwraps misfolded proteins, damaged organelles, and intracellular pathogens. Gradually the membrane completely surrounds these materials and converts into a structure called autophagosome. In the end, autophagosome fuses with a lysosome, which degrades their contents to recycle nutrients as point out in Figure 1. In addition, the internal membrane of the autophagosome is destroyed (71,88). Autophagy is considered as a nonspecific mechanism for degradation since recent studies have shown that several receptors identify the cargo specifically. These receptors include NBR1, OPTN, NDP52, NIX, and SQSTM1/p62 (3,4).

Autophagy is a process of bulk protein degradation that involves several sequential steps: at first, a double membrane isolated from the cytoplasm, trans-Golgi, or ER begins to enwrap misfolded proteins and damaged organelles, and gradually this double membrane is completed. In the next step, the autophagosome is formed when a double membrane is completely surrounding the misfolded proteins and damaged organelles. Finally, autophagosome fusion with lysosome destroys macromolecules and recycles them into cytosol. ER, endoplasmic reticulum.
HBV Induces ER Stress
In 1973, the first sign of the ER stress and the intracellular morphological change caused by HBV infection was identified, showing strong hypertrophy of the ER in hepatocytes using cirrhotic and carcinoma liver biopsies. Because of their microscopic appearance, cells were called “ground glass hepatocyte” (GGH) (42,96). During chronic HBV infection, the accumulation of immune-escape preS mutant surface antigens in the ER is linked to preneoplastic GGHs (16). Wang et al. (126) studied a total of 50 samples from eight resected liver specimens to confirm the types of GGHs harboring preS1 and preS2 mutations and demonstrated the activation of ER stress through different preS mutants using an in vitro model. They reported that type I GGHs had deletions in the preS1 region, as well as an aggregated pattern of hepatitis B surface antigens, whereas type II GGHs contain deletions over the preS2, specifically cytotoxic T cell binding epitopes. Expression of preS mutant large surface antigen in transgenic mice models induces GGHs and misfolded protein response or ER stress signals with activation of VEGF/Akt/mTOR and COX-2/NF-κB signals, causing oxidative DNA damage, aneuploidy, and genomic instability (20).
In addition, preS2-LHBs trigger calcium release from the ER and stimulate store-operated calcium entry (SOCE). SOCE increased ER and plasma membrane (PM) networks, which were connected through ER-resident stromal interaction molecule-1 (STIM1) protein and PM-resident calcium release-activated calcium modulator 1 (Orai1). Prolonged activation of SOCE culminates in chromosome aneuploidy, centrosome overduplication, aberrant multipolar division, anchorage-independent growth, and xenograft tumorigenesis in hepatocytes expressing preS2-LHBs (137).
HBV activates the ER-associated degradation (ERAD) pathway and leads to the ER degradation-enhancing mannosidase-like protein (EDEM) expression. Finally, it facilitates the degradation of HBV surface proteins through autophagy machinery as summarized in Table 1 (50,70).
Factors Involved in Regulation of Autophagy in Hepatitis B Virus Infection
The piling of misfolded or unfolded proteins in the lumen of the ER triggers ER stress. To alleviate the ER stress, cells recruit a complex signaling cascade named the unfolded protein response (UPR), the major ER stress pathway, which relies on activation of three complex signaling pathways: the activating transcription factor 6 (ATF6), the inositol-requiring enzyme 1 (IRE1), and the double-stranded RNA-activated protein kinase-like ER kinase (PERK) to stimulate downstream signaling to deal with misfolded or unfolded proteins (47,101). UPR will relieve ER stress by decreasing protein synthesis, ER chaperone protein upregulation to enable protein folding, and enhancing protein degradation through autophagy and ERAD pathway. These ER membrane-associated proteins act as autophagy inducers through the activation of the Signal Transduction Pathways, including ATF4, CHOP, NF-kB, eIF2a, and JNK1 (64). If the UPR remains unsuccessful to alleviate ER stress, it will induce apoptosis (73,92). Besides, ER stress release calcium from ER leads to mitochondrial deficiency connected to the overproduction of ROS. These stress signals cause oxidative DNA damage, mutagenesis, and autophagy induction (5,143).
The immune system produces ROS within the antiviral immune response trying to eliminate HBV-positive cells. HBV-triggered ROS production might contribute to viral pathogenesis to a considerably less extent in comparison with the ROS production induced through the host immune response because HBV-replicating cells activate the expression of cytoprotective genes by activating the Nrf2/ARE signaling pathway (83). The antioxidant response element (ARE) is a cis-acting enhancer sequence that intermediates transcriptional activation of genes in cells exposed to oxidative stress. The main regulator of ARE mediated gene expression is carried out using NF-E2-related factor 2 (Nrf2). The Nrf2/ARE is triggered by the HBV-regulatory proteins HBx and LHBs through c-Raf and MEK. Eventually, with the activity of Nrf2/ARE-regulated genes, the virus-infected cells are protected against immune responses and lead to virus replication. It has also been shown that Nrf2/ARE-regulated genes are more active in HCC and can protect tumor cells against the immune system (105).
It was suggested that the UPR-induced autophagy is essential for the effective envelopment of the HBV nucleocapsids. This result was based on the reduced secretion of enveloped virions from 3-MA treated cells and the increased enveloped virion titer in the supernatant of the cells exposed with autophagy activator like rapamycin and starvation (72).
Autophagy Crosstalk with HBV Replication
Sir et al. (110) showed that HBV induces early autophagy to enhance its DNA replication. It was suggested that early autophagy induction in ER stress may act as platforms for viral DNA replication and assembly. It has revealed that HBV induces autophagosome but inhibits complete autophagy formation. One of the factors that cause incomplete autophagy is HBV damp down Rab7 (a small GTPase) expression, which is responsible for the autophagosome-lysosome fusion (52,147). The impaired autophagy in HBV transgenic mice with liver-specific knockout of Atg5 caused a major reduction of both, HBV DNA and viral particle secretion in the mice sera (123). Döring et al. suggested the autophagic Atg5–12/16L1 elongation complex together with Atg10 and Atg3, which normally drive autophagophore membrane expansion, to be an important platform for HBV nucleocapsid assembly and stability. Inhibition of Atg5–12/16L1 and Atg10/Atg3 significantly reduced progeny virus yields. HBV through the interaction of its core protein with the Atg12 moiety of the complex gained access to Atg5–12/16L1. In contrast, HBV does not need LC3B or LC3/GABARAP (Gamma-aminobutyric acid receptor-associated protein precursor) lipidation and, consequently, does not depend on phagophore maturation and closure stages. The inessentiality of the late degradative steps of autophagy can elucidate why HBV activates autophagy without completion in the destructive autophagolysosomes (32).
HBV X protein (HBx) is a multifunctional oncoprotein involved in viral pathogenesis and carcinogenesis. HBx regulates a wide range of cellular genes in trans and has a versatile role in modulating transcription, protein degradation pathway, cell cycle progress, signal transduction, apoptosis, and genetic stability (118). HBx is quite unstable and is present at low levels in the host cell. HBx is retained at a very low intracellular level since it is rapidly degraded using proteasome complex, with a half-life of ∼30 min (58). Chang et al. (15) reported that low-level ectopic HBx expression in Huh7 cells triggers more significant autophagosome formation compared with high-level HBx expression. HBx under low-level expression activated beclin-1 promoter and upregulated the beclin-1 protein. Transcription factor AP-1 acts as an essential factor in HBx-mediated beclin-1 promoter activation. RhoA and its downstream effector Rho-associated coiled-coil-containing protein kinase 1 (ROCK1) inhibition significantly decreased HBx-induced autophagy. Transiently-expressed HBx stimulated an augmented RhoA-GTP level, as well as phospho-ROCK1 transient pile up (15).The promoter of the Beclin 1 gene is located in nt −277/+197 and exhibits the maximum promoter activity (118). Beclin 1 gene is the first and most important known genes involved in HBV infection and autophagy stimulation. HBx is the most prominent protein of HBV in the stimulation of autophagy. It has been shown that when the function of this protein is blocked by interfering RNA (siRNA), it indicates the autophagy dependent on this protein (106,118). Beclin 1 through an apoptotic modulator, death-associated protein kinase (DAPK), can be phosphorylated which in turn led to induce autophagy (141,142). HBx induced starvation-induced autophagy by increasing the activity of DAPK. In addition to DAPK, other pathways can also be involved in the regulation of autophagy. These pathways include different molecules such as mTOR, phosphatidylinositol-3-kinase (PI3K), and c-Jun N-terminal kinase (JNK) (80,128,142). HBx protein hinders autophagy degradation through disrupting lysosomal maturation, and this may be critical to the development of HBV-induced HCC (77). HBx induced autophagosome formation is occurred by reduced degradation of both LC3 and SQSTM1/p62. HBx interferes with lysosomal acidification leading to a deterioration of lysosomal digestive capacity probably due to mistrafficking of the V-ATPase involved in lysosome acidification. This leads to the accumulation of malfunctioning lysosomes containing immature hydrolases, which are important to degrade the cargo proteins (77,89). The formation of autophagy is mediated by its X protein through interaction and activation of phosphatidylinositol-3-kinase class 3 (PI3KC3) (110). The initiation of autophagy by HBV requires HBx, which binds to PI3KC3 to enhance its activity. Some studies have shown that the induction of autophagy in this way cannot ultimately lead to an increase in the autophagic protein degradation rate. In addition, inhibition of the hVps34 or Atg7 proteins by siRNA or PI3KC3 by 3-methyladenine has led to the suppression of HBV DNA replication (111). Li et al. (72) reported that deletion of the HBV envelope proteins disrupts the HBV-induced autophagosome formation. SHBs do not affect the expression level of Beclin 1 and, therefore, involves a different mechanism to that of HBx-induced autophagy (72). In Figure 2, a summary of the connection between the HBV and the autophagy mechanism is presented.
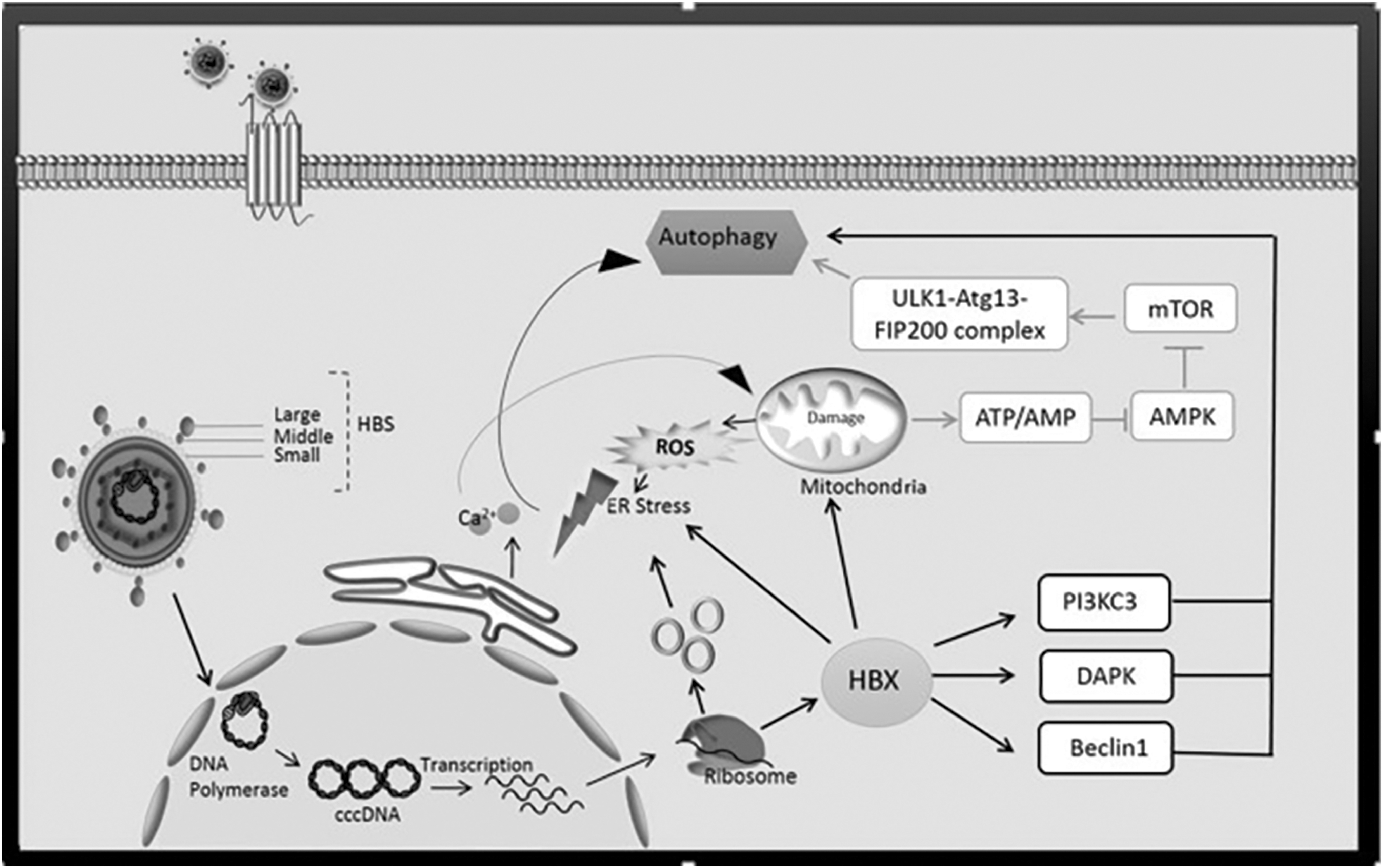
The HBV particles bind to various hepatocellular receptors, (such as transferrin, asialoglycoprotein, and other receptors) through S glycoprotein. This glycoprotein is involved in the fusion process, so S protein is involved in both the binding and the fusion (72). After entering the cell cytoplasm, the virus enters the cell nucleus using the importin alpha and beta. In the nucleus, the endogenous polymerase completes the positive strand and converts to a model for the synthesis of viral mRNAs. These mRNA transcripts enter into cytoplasm for translation to various proteins (99). HBx enhances autophagy with a positive effect on the Beclin 1 and leads to Beclin 1 phosphorylation through PI3KC3 and DAPK pathway (118). The Beclin 1 results in inositol trisphosphate receptor (IP(3)R) present in the endoplasmic reticulum, culminating in leakage of Ca(2+) from the endoplasmic reticulum in which the autophagy will be activated. Under these conditions, calcium released from the ER stimulates impaired mitochondrial activity connect to excessive production of ROS. In addition, released Ca2+ raises ATP production (AMP/ATP) from the mitochondria which in turn blocks the AMPK mTOR and ULK1-Atg13-FIP200 complex. In addition, the C-terminal of HBx is required to produce ROS. The ROS production causes DNA damage and promotes DNA mutation. Especially, mitochondrial DNA is more sensitive to mutation and cleaned by mitophagy. The ROS along with X and S proteins causes stress in the endoplasmic reticulum, which is involved in direct stimulation of autophagy (55). ROS, reactive oxygen species; HBV, hepatitis B virus.
Double Role of Autophagy in HBV-Related Tumorigenesis
In cancer, autophagy has paradoxical roles, acting both as a tumor suppressor in the early stage of cancer development and as a tumor promoter in the end stage of cancer (6). Autophagy protects cancer cells from several stress conditions, such as hypoxia, starvation, oxidative stress, and chemotherapy (3). Extensive evidence suggests that 50–60% of cancers increase under hypoxic conditions in which autophagy will be triggered (130). Autophagy through activation of the epithelial–mesenchymal transition increases HCC invasion (74). In addition, autophagy with a negative effect on p53 increases HCC (102). Gong et al. (37) presented that autophagy stimulates the growth of cancerous stem cell-derived mammary tumors. In addition, autophagy deficiency can increase instability in chromosomes, the accumulation of defective cells and misfolded proteins, generation of ROS, induction of oxidative stress, and increasing DNA damage (24,134). In contrast, a large collection of articles implicating autophagy may inhibit tumorigenesis of HBV-associated HCC (132). In the HBx transgenic mice model with late stages of liver tumor, autophagic activity is diminished following liver tumor formation. Qu et al. (98) revealed that the heterozygous deletion of Beclin 1 in the HBV transgenic mice leads to malignancies and accelerated HBV-induced HCC spontaneously. Deletion of ATGs UVRAG increased susceptibility to HCC development in mice (75). In HBV-associated HCC, autophagy was downregulated and there was a reverse relationship between the autophagic activity and the level of microRNA-224 (miR-224) in these tumor cells (68). Autophagosomes can sequester miR-224 and subsequently degrade it. The increase of miR-224 results in the downregulation of its target gene Smad4; a transcription factor is required for the activation of the TGFβ signaling pathway (68). Smad4 inhibition could transform TGFβ from a tumor suppressor to a tumor enhancer (140). In a clinical investigation, Kotsafti et al. (63) exhibited that the mRNA of Beclin 1 was significantly lower in HCC tissues than in chronic hepatitis tissues. In Figure 3, a comparison has been made between the role of autophagy as a tumor suppressor and tumor promoter.
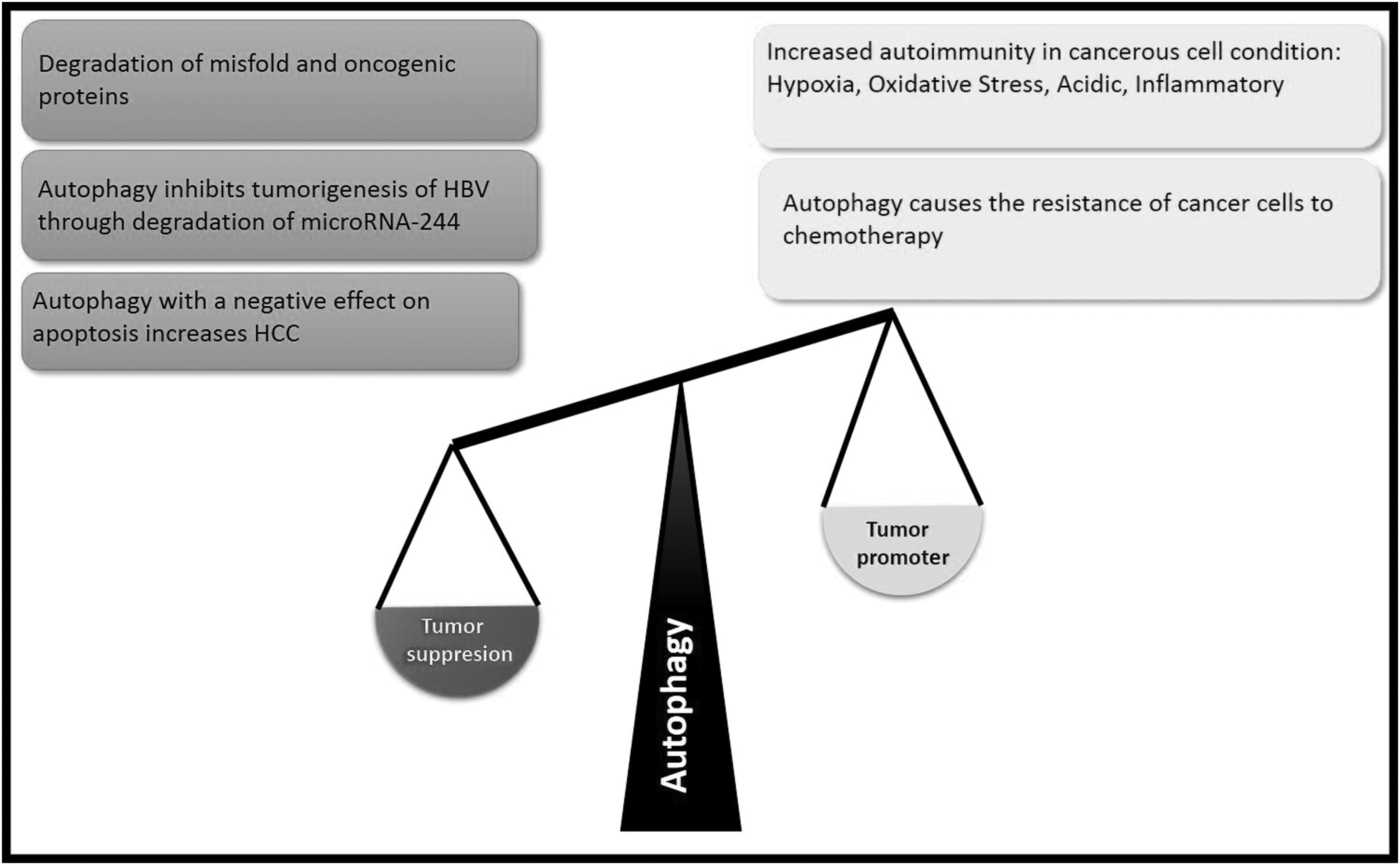
In this figure, a comparison was made between the role of autophagy as a promoter or suppression in the tumor. The suppression roles of autophagy in the HCC include the degradation of misfolded and oncogenic proteins and inhibition of tumorigenesis through degradation of microRNA-224. But autophagy may work on in favor of cancer. In the end stage of cancer autophagy helps the cancer cell survival. In addition, autophagy helps the resistance of cancer cells to chemotherapy especially in hypoxia, oxidative stress, acidic, and inflammatory conditions. HCC, hepatocellular carcinoma.
Signaling Pathways Involved in HBV-Related Autophagy
Mitogen-activated protein kinase (MAPK) pathways are involved in autophagy induction (148). Three pathways of MAPK, that is, extracellular signal regulated kinase (ERK), p38 MAPK, and c-Jun N-terminal kinase (JNK), were triggered by HBx. HBx mediated phosphorylation of Bcl2 will be stopped if JNK signaling is inhibited. It causes dissociation of Beclin1 from Bcl2, a step important for autophagy initiation (146). Adenosine monophosphate-activated protein kinase (AMPK) is crucial in bioenergetic homeostasis to preserve cellular adenosine triphosphate (ATP) (87). Definitely, some pathogens can modulate the activity of AMPK/mTOR to get sufficient energy for their proliferation and growth (13). In HBV infection, AMPK can support or hinder viral replication. Xie et al. (133) suggested that AMPK activated through HBV-induced ROS accumulation can inhibit HBV replication. Mechanistically, AMPK activation culminates in HBV-mediated autophagic activation, which increases autolysosome-dependent degradation to limit viral proliferation (133). HBx plays significant roles through activating other signal pathways such as Ras–Raf, PI3K-Akt/PKB, PI3K/Akt, and PI3K/Akt/mTOR (112,119). HBx prevents apoptosis by acting as a caspase 3 inhibitor (38), inhibiting the Fas-mediated apoptosis (30), hindering the initiation of apoptosis through the formation of the survivin-HBXIP complex, and preventing procaspase 9 (144). HBx directly interacts with p53 and blocks it, downregulates the expression of PTEN and activated Akt (19), as well as by reducing the production of S-adenosyl-methionine and upregulation of MAT2A through NF-KB and cAMP–response–element–binding protein (CREB) signaling pathways (78) leads to inhibition of hepatoma cell apoptosis. In contrast, HBx induces apoptosis by interacting with signaling proteins such as c-FLIP (59) and HSP60 (104), increasing FAS-L expression (61) and interacting with Bax .(43) The role of HBx in inhibiting or inducing apoptosis depends potentially on the situation. HBx in acute HBV infection induces apoptosis and in chronic infection and HCC cells inhibits apoptosis.
Cytokines Involved in HBV-Related Autophagy
A growing body of evidence suggests that autophagy suppresses inflammation. Several studies have proven that the autophagy induction by the HBx increases pro-inflammatory cytokines such as IL6, IL8, and CXCL2. This protein also induces other cytokines such as tumor necrosis factor α (TNFα), transforming growth factor β (TGFβ), interferon γ inducible protein-10 (IP-10), and nuclear factor-κB .(82) High level of IL-6 can predict the progression of HCC disease in a person with chronic hepatitis .(131) Similar to IL-6, elevated level of IL-8 has been observed in people with chronic HBV with active inflammation .(121) IL-8 has also been demonstrated to decrease the antiviral activity of interferon-α against HBV (95). CXCL2 overexpression has been shown in hepatic inflammation caused by chronic alcohol consumption .(82) TGFβ has an important function in autophagy. TGFβ cytokine increases the autophagosome formation, stimulating the expression of the mRNA expression levels of Beclin 1, ATG5, ATG7, and DAPK (45,53). Studies showed that HBx induces autophagy through activating DAPK in a pathway related to Beclin 1, but not JNK. Although HBx could activate JNK, the change in JNK failed to influence HBx-induced autophagy (142). Usually, TGFβ stimulates the autophagy pathway through the Smad (Smad2/3, Smad4) pathway. It has also been proven that TGFβ Cytokine stimulates autophagy earlier than apoptosis (62). Interferon γ is an important and multifunctional cytokine, and its important properties are its antiviral performance. This cytokine stimulates the autophagosome formation through IRF-1 signaling pathway. In addition, interferon γ in HCC causes autophagy to inhibit cell growth and death. In HBV infection, protein X enhances TNFα .(69) It has been shown in many studies that TNFα promotes the autophagy in the context of HBV infection through enhancing the expression level of LC3 and Beclin 1.
HBx protein sensitizes cells to apoptotic killing by TNFα. This induction of apoptosis is done by prolonged stimulation of N-Myc and the stress-mediated mitogen activated-protein kinase 1 (MEKK1) pathway but not by upregulating TNF receptors (114). In addition, HBx by activation of NF-κB can block TNFα and FAS-mediated apoptosis pathway (93).
Interface Between the Autophagy and Different Types of Cell Death in Hepatocytes
The three major mechanisms that trigger programmed cell death are named type I (apoptosis), type II (autophagic cell death), and type III (Necroptosis) (109). Autophagy displays bidirectional functions in cell destiny determination based on the intensity and duration of inducers (136). Normally, autophagy is executed at a basal level in every cell and stimulates cellular homeostasis against cellular stressors and organelle turnover (79). Under certain stress conditions, autophagy also can be utilized as a cell-suicide mechanism (108).
Type II cell death or autophagic cell death is induced in apoptosis-resistant cells, mainly in the lack of the pro-apoptotic proteins Bax and Bak and caspase-independent manner. The term autophagic cell death can be used when cell death is blocked through the inhibition of autophagy by chemical inhibitors such as 3-methyladenine and wortmannin or genetic manipulation like knockout or siRNA silencing of essential autophagy genes (109). Interconnection between autophagy and other cell death types has a vital effect on the fate of the hepatocytes. Cell death is a central mechanism of liver injury that can be attributed to a wide array of causes such as acute and chronic viral hepatitis, alcoholic and nonalcoholic steatohepatitis, drug-induced liver injury (33), cholestasis, steatosis, and autoimmunity (127). Pietro Di Fazio et al. (29) reported that panobinostat (histone deacetylase inhibitors [DACi]) hyperactivates autophagy formation in HCC cells and combines with a broad spectrum of mechanisms to promote autophagic cell death. Now, autophagy-mediated cell death represents a novel approach to combat against cancers, particularly for tumors that show resistance to apoptosis.
The p53 and its family members play a critical role in autophagy. Despite its recognized role as guardian of the cell fate through the control of the apoptotic pathway, p53 through promoting a pro-survival or pro-death autophagic machinery exerts a double-edged role during autophagy (103). Tumor suppressor p53 protein can co-regulate autophagic cell death and apoptosis. The p53 modulates the expression of pro-apoptotic protein such as Apaf1 and Bcl-2 and autophagy-related pathways, including Bmf/Beclin-1 and AMPK/mTOR. In addition, p53 targets the expression of DNA damage-regulated autophagy modulator 1 (DRAM) and can stimulate both autophagic cell death and apoptosis as demonstrated in Figure 4 (103). Generally, increasing the nuclear p53 after DNA damage leads to the induction of autophagic cell death through phosphorylation and activation of AMPK and JNK1 (54,64). In contrast, increasing the cytoplasmic p53 inhibits autophagic cell death and induces apoptosis (39,120).
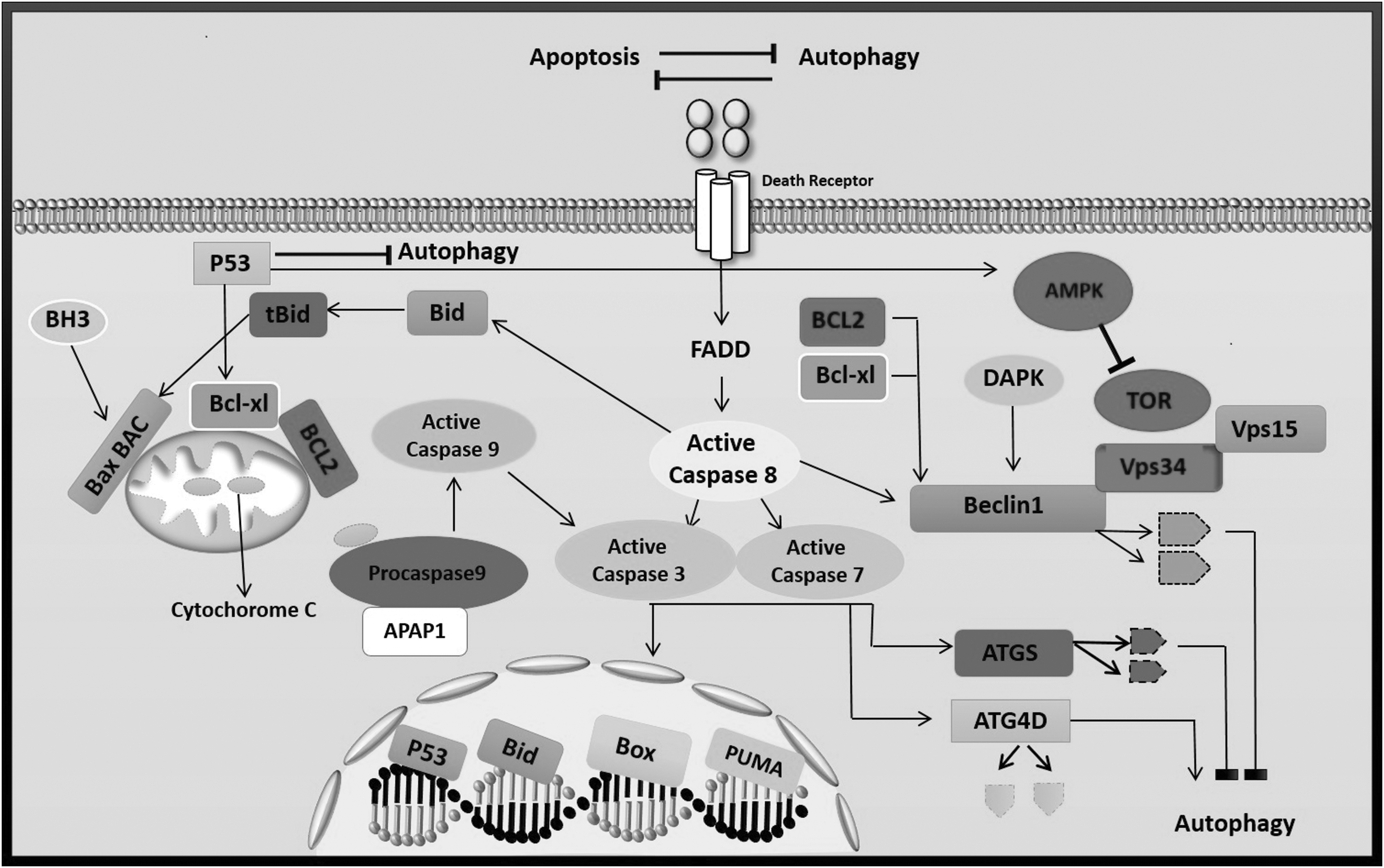
The molecular crosstalk between autophagy and programmed cell death. Beclin 1, one of the most important molecules in the autophagy pathway along with two other molecules (VPS34 and VPS15), forms a complex that plays an important role in the formation of the autophagosome platform. Two antiapoptotic molecules (BCL-2 and BCL-XL) with a negative effect on Beclin 1 prevent autophagy (51). In addition, molecules of caspase with effect on the Beclin 1 and ATG5 cause them to break down and lose their ability to stimulate autophagy. Although apoptosis-associated cleavage of Beclin 1 and Atg5 disables autophagy, the cleavage of Atg4D through caspase-3 generates a fragment with enhanced autophagy activity (86). Thep53 communicates with autophagy in different ways. Increasing the nuclear p53 triggers a range of genes that have a positive effect on autophagy. Specially, the proteins are involved in nutrient sensing pathways and act as mTOR regulator (AMPK, TSC2, and SESN2). In addition, p53 triggers other genes which are involved in autophagy regulation. These genes include DRAM1, ISG20L1, DAPK-1, BAX, and PUMA. They have roles in cell death. Low contents of p53 are also showed to negatively regulate autophagy in the cytoplasm in the absence of p53 target gene activation. This role has also been shown for tumor-resulting mutants of p53 (21,103).
Two antiapoptotic proteins Bcl-xL and Mcl-1 from the Bcl-2 family suppress apoptosis by blocking the activation of the intrinsic apoptosis pathway by interacting with the pro-apoptotic molecules (like Bax) (14). The specific deletion of each of these antiapoptotic proteins results in apoptosis and elevation of serum ALT and hepatic fibrosis (49,117). Bcl-2/Bcl-xL can also interact with the Bcl3 domain of Beclin 1 and thereby hinder autophagy (94).
Apoptosis is a chronic and moderate response after damage stimulation, while necrosis is an acute and severe response. Sustained apoptosis in hepatocytes is often accompanied by fibrogenesis, chronic liver function impairment, and even cancer development (84). For attenuating the virus-induced apoptosis, HBV/HBx induces the mitochondrial fission (fragmentation) and mitophagy (selective degradation of mitochondria by autophagy) through a perinuclear clustering of damaged mitochondria and translocation of the dynamin-related protein (Drp1) to mitochondria by stimulating its phosphorylation at Ser616. HBV also stimulated the expression of Parkin, an E3 ubiquitin ligase, PINK1, and LC3B translocation, and self-ubiquitination of Parkin in the mitochondria facilitates the ubiquitination and degradation of its mediator of mitochondrial fusion named as Mitofusin 2 (Mfn2), which leads to mitophagy (17,60).
Apoptosis, necroptosis, and necrosis may exist side by side in acute and chronic liver disease (81). Necrosis is an “immunogenic” form of cell death that is characterized by mitochondrial impairment, depletion of ATP, and subsequent failure of ATP-dependent ion pump. Necrosis initiates with increasing Ca++ and ROS in the cell, and necroptosis initiates upon tumor necrosis factor receptor (TNFR) activation. Indeed, Necroptosis is a programmed form of necrosis. In contrast to necrosis, leaking of the membrane is regulated by the cell during necroptosis. Both of these forms of cell death ultimately lead to rapid swelling of cells and cell organelles (“oncosis”), spilling into the extracellular environment, and elicit significant inflammatory responses (36,56). In hepatocytes treated with acetaminophen (APAP), damaged mitochondria can be cleaned by canonical mitophagy resulting in reduced necrosis (22).
The autophagic machinery may directly crosstalk with apoptotic factors or necrotic pathways to enhance cell death. For example, Atg5 has been reported to interact with FADD (Fas-associated protein with death domain) and stimulate caspase-dependent cell death (97). Another example is that Atg5 can be cleaved by the action of calpains (138,139). This cleaved form will be translocated at the level of the mitochondria and interact with Bcl-xL to remove its inhibition on Bax. The activation of Bax allows the mitochondrial outer membrane permeabilization and the release of pro-apoptotic factors, including cytochrome c. After the association of cytochrome c with apoptotic protease activating factor 1 (Apaf-1) and forming of the apoptosome, autoactivated procaspase 9 cleaves and activates caspases 3 and 7 that finally degrade of cellular substrates and culminate in apoptosis (138). Finally, Bcl-2/Bcl-xL can interact with the Bcl3 domain of Beclin 1 and thereby hinder autophagy (94).
Autophagy-Related Potential Therapy for HBV Infection and HCC
Currently, chemotherapy is almost ineffective for HCC due to chemoresistance and limitation of liver function (129). Autophagy is identified to promote cancer resistance to chemotherapy. Autophagy inhibition using 3-MA or the siRNA targeting Beclin-1 improved chemotherapy-induced apoptosis by causing changes in mitochondrial membrane permeability and structure (41). Rapamycin is an autophagy inducer agent which by inhibiting mTOR pathway causes to improve overall survival rates in post-liver transplantation in patients with HCC (124). Sirolimus (autophagy inducer) in patients with advanced HCC who did not have liver transplantation has similar results (26). Sorafenib is an FDA-approved drug that activates autophagy through inhibition of the mTOR pathway, which improves the survival of patients with advanced HCC in preclinical studies and several clinical trials (18). Exposure of cancer cells with pan-HDI (HDAC inhibitors), for instance, vorinostat and panobinostat, is recognized as autophagy inducer in cancer cells, particularly if caspase activity or apoptosis induction is blocked (107). Treatment with panobinostat, a pan HDAC inhibitor, combined with sorafenib has been shown the highest efficacy in HCC (67). The combination of sorafenib with SAHA (Vorinostat is also known as suberanilohydroxamic acid) has promoted responses against HCC compared to treating with sorafenib alone (67). Autophagy can selectively degrade lipid to protect hepatocytes against alcohol-induced hepatotoxicity and steatosis (31). Autophagy induction can be considered as a generalized treatment against steatosis; nevertheless, the upregulation of autophagy in hepatic stellate cells elevates their activation and subsequently initiates liver fibrosis (122). In contrast, inhibition of autophagy can result in the accumulation of lipid droplets in the cytoplasm of stellate cells, leading to apoptosis of hepatic stellate cells (46). Using autophagy inducer agents such as carbamazepine (CBZ) and rapamycin was reported to reduce hepatic steatosis in alcoholic fatty liver disease and nonalcoholic fatty liver disease and most likely connected to the augmentation of autophagy, in turn, leading to degradation of lipids (76). One method of controlling HCC tumorigenesis is to induce hepatoma cell death through autophagy formation. .(65) Zhong et al. (145) indicated that there is a substance called Epigallocatechin-3-gallate (EGCG) in tea that is involved in the replication of the HBV by the mechanism of autophagy. This substance causes to complete formation of autophagy in hepatoma cells, thus preventing HBV replication. Su et al. (113) reported that soybean fermentation products containing live bacteria were used to inhibit liver tumorigenesis through the activation of apoptosis and autophagy without significantly changing the mean body and liver weight in a syngeneic mouse model. It has been revealed that CBZ as autophagy inducer could decrease the α-1 antitrypsin load and risks of liver fibrosis and cancer in an α-1 antitrypsin deficiency mouse model suggesting that autophagy activation might provide preventive effects against liver cancer (48). mir-141 can suppress HBV replication through targeting of Sirt1 (bound to the 3′UTR), which plays an important role in regulating the autophagy by deacetylation of autophagy-related proteins (Atg5, Atg7, Atg8) (100,135). Furthermore, mir-141 targets microtubule-associated protein light chain 3 (LC3). LC3 is a crucial protein in the autophagy pathway where it plays a role in autophagy substrate selection and biogenesis of autophagosome. The maturation of LC3 and the conversion of LC3I to LC3II in HBV were decreased by mir-141 (27,135).
Perspective
As aforementioned, the association of the virus with autophagy can increase the virus's replication, so interfering with this relationship can achieve different therapeutic approaches which are confirmed by the use of EGCG in tea and some medicines (145). Of course, this therapeutic approach should be such that it does not prevent the complete stimulation of autophagy, because to prevent autophagy, conditions for other infections, such as cancer (HCC), are provided (145).
Abdoli et al. reported that autophagy controls virus replication in a time-dependent manner in the case of HCV and Influenza virus (2,34). Furthermore, they showed that autophagy induction and inhibition in prophylactic (before virus infection) and therapeutic approach (after virus infection) have different outcomes in virus replication. From this point of view, autophagy manipulation through vaccination or in combination therapy may pave a new way to combat viral hepatitis. Selectively targeting antigens to autophagy machinery may be a promising approach to elicit CD4+ and CD8+ T cell responses. Our team used Beclin-1 as an adjuvant with HEV candidate vaccine in the animal model, and the results indicated that the groups that received the Beclin-1 formulated vaccine raised immune responses significantly (57,90,91).
From other therapeutic approaches, the drug resistance should be pointed out. The autophagy induction culminates in the downregulation of nucleoside transporters, equilibrative nucleoside transporter 1 (ENT1) and concentrative nucleoside transporter1 (CNT1). Low expression of ENT1 or CNT1 has been linked to nucleoside drug resistance (1). In addition, autophagy activation is able to suppress interferons, which leads to drug resistance as reported in the HCV virus infection context. A combination of autophagy inhibitor agents with HBV and HCV drugs may lead to reduction of drug resistance and increase of drug efficacy.
Telomerase activity has been proven to increase tumor cell survival, and telomerase activation is associated with HBx in HCC. We showed that the stimulation of autophagy reduces the activity of telomerase (116). Therefore, these results can be used to propose a therapeutic strategy that using autophagy stimulating agents may reduce HBV linked HCC.
Conclusion
Hepatocytes are believed to have higher levels of autophagy compared with other cell types owing to their abundance of lysosomes and lysosomal enzymes. Autophagy has primarily been regarded as having a hepatoprotective and antitumor role in normal liver cells by maintaining cell homeostasis during liver injury. But a more complex scenario is emerging with the recognition of the pro-fibrogenic, pro-tumor, and proviral role of autophagy in HBV infected cells. A deep understanding of the mechanisms in which autophagy displays versatile roles at different stages of HCC and HBV infection is urgently needed. As autophagy is a ubiquitous cellular event, exploration of autophagy and HBV crosstalk and development of effective drugs to manipulate autophagy in hepatocytes may be a great hotspot and milestone for decreasing HCC risk in chronic HBV infection and improving HCC therapeutic efficacy.
Footnotes
Acknowledgment
The authors thank the staff of the Hepatitis and AIDS department, Pasteur Institute of Iran, for kind assistance and support.
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
Funding Information
This work was supported by research grant number 940 from Pasteur Institute of Iran.