
Abstract
Immune regulation at the maternal-fetal interface is complex due to conflicting immunological objectives: protection of the fetus from maternal pathogens and prevention of immune-mediated rejection of the semiallogeneic fetus and placenta. Interferon (IFN) signaling plays an important role in restricting congenital infections as well as in the physiology of healthy pregnancies. In this review, we discuss the antiviral and pathogenic effects of type I IFN (IFN-α, IFN-β), type II IFN (IFN-γ), and type III IFN (IFN-λ) during pregnancy, with an emphasis on mouse and non-human primate models of congenital Zika virus infection. In the context of these animal model systems, we examine the role of IFN signaling during healthy pregnancy. Finally, we review mechanisms by which dysregulated type I IFN responses contribute to poor pregnancy outcomes in humans with autoimmune disease, including interferonopathies and systemic lupus erythematosus.
Introduction
Immune regulation at the maternal-fetal interface is complex due to conflicting immunological objectives: protection of the fetus from maternal pathogens and prevention of immune-mediated rejection of the semiallogeneic fetus and placenta. Maternal and fetal tissues are in direct contact where the fetal-derived placenta invades and implants into a layer of specialized maternal endometrial tissue, termed the decidua. The decidua is enriched in maternal leukocytes, particularly natural killer (NK) and Treg cells, which mediate protective and tolerogenic immunity at the maternal-fetal interface. In addition to controlling pathogen transmission from mother to fetus, proper regulation of cytokine signaling, including interferon (IFN) signaling, contributes to the physiology of healthy pregnancy.
In this review, we discuss the antiviral and pathogenic effects of type I, II, and III IFN signaling during pregnancy, with an emphasis on mouse and non-human primate (NHP) models of congenital Zika virus (ZIKV) infection. In the context of these animal model systems, we examine the role of IFN signaling during healthy pregnancy. Finally, we review mechanisms by which dysregulated type I IFN responses contribute to poor pregnancy outcomes in humans with systemic autoimmunity.
IFN Signaling
IFNs are cytokines and are divided into three families (type I, type II, and type III) based on sequence homology, evolutionary relatedness, receptor usage, and functional activity
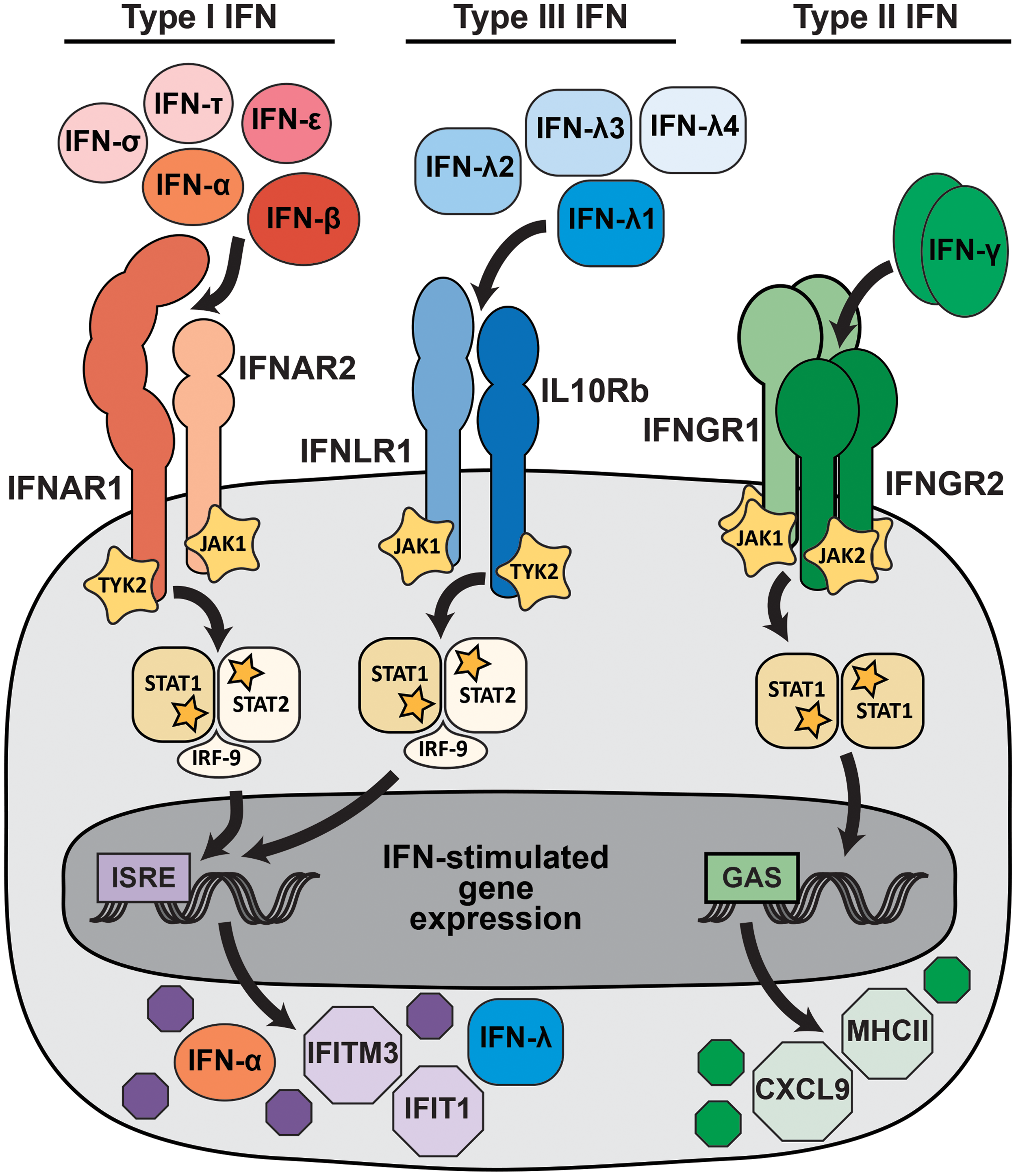
IFN signaling pathways. Type I, type II, and type III IFNs signal through distinct receptors, but activate overlapping transcriptional programs. More than a dozen type I IFNs, including IFN-α, IFN-β, IFN-ɛ, IFN-δ, and IFN-τ, signal through a heterodimeric receptor comprising IFNAR1 and IFNAR2. A smaller set of type III IFNs (IFN-λ) signal through a heterodimeric receptor comprising IFNLR1 and IL10Rb. The type II IFN family consists only of IFN-γ, which signals as a dimer through a tetrameric receptor comprising IFNGR1 and IFNGR2. Receptor binding activates kinases, including JAK1, JAK2, and TYK2, which phosphorylate STAT1 and STAT2. Phosphorylated STATs dimerize and translocate to the nucleus, where they activate transcription from promoters that contain ISRE (STAT1/2 heterodimers) or GAS (STAT1 homodimers), resulting in the expression of IFN-stimulated genes. The canonical IFN signaling pathways are depicted, but additional signaling pathways also are activated and likely contribute to the specific transcriptional response induced by different IFNs. GAS, γ-activated sites; IFN, interferon; ISRE, IFN-stimulated response elements. Color images are available online.
Despite using different receptors, type I and type III IFNs share many functional similarities. Both are induced by pattern recognition receptor (PRR) signaling following viral infection, their receptor binding activates STAT1/STAT2 heterodimer formation, and they induce a similar transcriptional program characterized by IFN-stimulated response elements in the promoters of induced genes (5,61,68). Type I and III IFN signaling result in the upregulation of hundreds of IFN-stimulated genes (ISGs), many of which act in a cell-intrinsic manner to restrict viral entry, replication, and spread (101). In addition to these canonical IFN signaling pathways, additional signaling pathways also are activated and likely contribute to the specific transcriptional response induced by different IFNs (61,87). Type I and III IFNs also serve immunomodulatory functions, including priming adaptive immune responses.
Although type I and III IFNs activate similar antiviral transcriptional responses, they have distinct physiological activities largely determined by receptor expression. IFNAR is ubiquitously expressed, but IFNLR expression is greatest on epithelial cells and some immune cell subsets, including neutrophils and NK cells (61). Furthermore, while type I and III IFNs are induced by similar PRR signaling pathways, some stimuli favor production of type III IFNs over type I IFNs. These stimuli include infection of respiratory epithelial cells (19,36,50,82), signaling by the PRR MAVS from peroxisomes rather than mitochondria (80), and signaling by the PRR TLR4 from the plasma membrane rather than endosomes (81). Overall, type III IFN signaling is less potent and less inflammatory than type I IFN and predominates at anatomic barriers, leading to a model wherein type III IFNs provide front-line protection at barrier surfaces, thereby minimizing the activation of the systemic type I IFN response and consequent immune pathology.
While type I and III IFNs are best known for their antiviral activities (61), type II IFN has distinct proinflammatory and immunomodulatory functions that also contribute to the control of viral infections. Type II IFN signaling induces STAT1 homodimers and induced genes are characterized by γ-activated sites (GAS) in their promoters. Type II IFN is produced by T cells and NK cells and functions primarily in later stages of infection by clearing viruses from infected tissues. Crosstalk between the type I and type II responses can impact the outcome of infection. For example, downregulation of IFNGR by type I IFN signaling leads to more severe Listeria monocytogenes infection in Ifnar1 −/− mice (95). Indeed, while Ifnar1 −/− mice generally have enhanced susceptibility to most viral infections, these mice are protected from infection by many bacterial and protozoan pathogens for which immune control is highly dependent upon type II IFN (13,97).
IFN Signaling During Pregnancy
Pregnancy encompasses multiple developmental stages, including implantation, fetal growth, and parturition, each with unique immunological requirements that have been reviewed elsewhere (73,75,121). Early events in pregnancy, such as blastocyst implantation and subsequent placental invasion, are inflammatory processes that physically degrade and remodel maternal tissue at the site of implantation. In contrast, fetal growth occurs in an anti-inflammatory Th2-type environment that characterizes the majority of pregnancy. Finally, parturition is an inflammatory process with NF-κB signaling contributing to labor induction. Since implantation, fetal growth, and parturition have distinct immunological features, IFNs play different roles at each stage of pregnancy. Accordingly, IFNs may also have distinct effects during viral infection at different gestational stages.
Before implantation, the blastocyst is surrounded by an outer trophectoderm layer that attaches to the maternal endometrium and differentiates into trophoblast layers that constitute the placenta. In mice, type I IFNs are expressed in the trophectoderm of the preimplantation blastocyst, in the decidua following implantation, and in multiple trophoblast layers mid-gestation (115). Accordingly, ISGs, including Irf-8, Iarp, Isg12, and Isg15, are upregulated in the postimplantation decidua (53). IFN-γ also is produced by human trophoblasts early in gestation and in mouse trophoblast giant cells, spongiotrophoblasts, and labyrinth zone by mid-gestation (84,90). Specialized NK cells (CD56bright and CD16−) with reduced cytotoxic potential, termed decidual, uterine, or endometrial NK cells, make up the majority of leukocytes in the maternal decidua and secrete IFN-γ during pregnancy (8,37). IFN-γ signaling at the maternal-fetal interface not only promotes differentiation of decidual NK cells but also facilitates formation of the placenta and maintenance of the decidua (75,121).
In addition to its role promoting the physiology of healthy pregnancy, type II IFN has been associated with adverse fetal outcomes in mouse models of congenital infection. Fetal loss following Toxoplasma gondii and Plasmodium berghei infection is ameliorated in Ifngr1 −/− mice (79,103). Likewise, administering an anti-IFN-γ antibody protects dams from fetal loss following Brucella abortus infection (56). Since IFN-γ is a key component of proinflammatory immune responses, fetal loss may be general response to infection and inflammation at the maternal-fetal interface in mice.
The placenta mediates nutrient and waste exchange between mother and fetus as the site of contact between fetal and maternal blood supplies. Maternal blood is delivered to the placenta by spiral arteries, which are remodeled early in pregnancy in a process involving IFN signaling. Mice lacking either type I or type II IFN signaling (Ifng −/−, Ifngr1 −/−, Ifnar1 −/−, or Stat1 −/−) have incomplete spiral artery remodeling, suggesting that both type I and II IFNs contribute to this process (75,117). Nonetheless, these knockout mice all produce healthy pregnancies and can be bred for routine experimental use.
Type I IFNs serve distinct pregnancy functions in other mammals. Ruminants express multiple subtypes of IFN-τ, which signals through IFNAR and induces ISG expression, but is not known to serve a protective antiviral function (31,57). Unlike IFN-αβ, IFN-τ expression is not induced by viral infection, but rather by trophoblast development (33). In trophoblasts, IFN-τ serves as a pregnancy recognition factor that modulates maternal hormonal status before implantation and induces ISGs in the maternal endometrium (43,98). In swine, another type I IFN, IFN-δ, is secreted by peri-implantation conceptuses and serves along with IFN-γ to modulate maternal endometrial gene expression before trophoblast attachment (38,67,100). Primates and rodents do not encode orthologs of IFN-τ or IFN-δ (57), which may reflect their corresponding placental types: ruminant and swine placentas are epitheliochorial and less invasive than the hemochorial placentas of primates and rodents (96). Although congenital ZIKV infection has been studied in swine (23,109,113), these studies have used in utero inoculation and thus do not model transplacental transmission.
IFN-ɛ, a type I IFN, is conserved in many mammals, including mice, humans, and NHPs, and is secreted constitutively in the female reproductive tract (27,35,44). In mice, IFN-ɛ is expressed primarily in the uterine endometrium, ovaries, and cervix, and protects against sexually transmitted infections, including HSV-2 and Chlamydia muridarum (35). However, IFN-ɛ levels fluctuate with the estrus cycle rather than being induced downstream of PRR signaling (27). In rhesus macaques, IFN-ɛ is secreted from mucosal epithelial cells in the vagina and cervix, as well as in the lung, foreskin, and small and large intestines (27). A role for IFN-ɛ during pregnancy has not been described, but its expression profile in the female reproductive tract potentially could protect the fetal compartment from ascending infections.
IFN Signaling in Mouse Models of Congenital ZIKV Infection
The 2015–2016 ZIKV outbreak throughout Latin America and the Caribbean led to the discovery that ZIKV infection during pregnancy can produce adverse fetal and neonatal outcomes, including microcephaly, intrauterine growth restriction (IUGR), and vision and hearing loss, as well as miscarriage. Subsequently, models of congenital infection were developed to test vaccines and antivirals as well as to define ZIKV pathogenic mechanisms and antiviral immunity at the maternal-fetal interface (85,88).
Mice have become a common animal model of congenital ZIKV infection (16). Comprehensive comparisons between mouse and human pregnancy have been reviewed elsewhere (6). Like humans, mice have hemochorial, discoid placentas and are an important pregnancy model because they are genetically tractable, cost-effective, and have a short gestation. However, modeling congenital ZIKV infection in mice can be difficult due to physiological differences at the maternal-fetal interface and because their gestational development has inexact parallels with human gestation. The shortened timing of mouse gestation (3 weeks compared to 40 weeks in humans) confounds studies of infection at distinct developmental stages as well as studies of adaptive immune responses during pregnancy. Furthermore, conventional inbred mouse models produce fetuses and placentas that are genetically identical to the dam; this removes the immunologic pressure of supporting invasive, semiallogeneic tissues that exist in outbred models or in humans.
Another complication of mouse models of congenital ZIKV infection is that type I IFN signaling restricts ZIKV replication in mice (59), in part, due to the inability of ZIKV to antagonize murine STAT2 and STING (28,40,41,58). This results in diminished ZIKV replication in immune-competent mice and has led most groups to use IFN-deficient mouse models, including mice lacking the type I and/or type II IFN receptors (e.g., Ifnar1 −/− or Ifnar1 −/− Ifngr1 −/− double knockout), mice with defects in IFN induction or signaling (e.g., Irf3 −/− Irf5 −/− Irf7 −/− or Stat2 −/−), or mice treated with IFNAR1-blocking antibody (74). Congenital ZIKV infection has been studied using IFN-deficient mouse models in which maternal ZIKV infection by intravaginal or subcutaneous footpad inoculation results in viral transmission to the fetus, placental damage, and adverse pregnancy outcomes, including IUGR and fetal demise (16,49,51,70,119). IFN-deficient mouse models are valuable because they allow sufficient viral replication to elicit congenital phenotypes, but they limit studies of the specific effects of IFN signaling at the maternal-fetal interface in the context of congenital ZIKV infection. Accordingly, some groups have reported congenital ZIKV infection in wild-type mice, although these models have required high inoculation doses, infection-enhancing antibodies, or direct intrauterine inoculation (22,94,107,110). ZIKV transplacental transmission (but not fetal pathology or loss) also has been reported in transgenic mice expressing human STAT2 (40). Recombinant IFN-λ2 administered to pregnant dams restricted ZIKV transplacental transmission (49) and induced ISGs in both placental and decidual cells (12), consistent with an antiviral effect of type III IFN at the maternal-fetal interface.
The mechanisms by which ZIKV accesses the fetal compartment have yet to be determined and could be affected by the route of inoculation (e.g., subcutaneous vs. intravaginal). Congenital infection also may be impacted by viral strain, as different ZIKV strains produce different pathologic outcomes in nonpregnant mice (59,106,108). Nonetheless, the ability to cause fetal infection and pathology in mice is not a unique property of contemporary ZIKV strains compared to historical ones (48,104,110). Infection at early gestational stages (before E10) results in higher rates of fetal loss and more severe IUGR (49,70,110,119), likely because placentation is incomplete and the placental and fetal tissues are more vulnerable to infection early in development. Fetal and placental pathology generally are more severe in Ifnar1 −/− dams compared to wild-type dams treated with an IFNAR1-blocking antibody, consistent with higher viral burdens in Ifnar1 −/− dams and suggesting that fetal disease outcomes are driven by maternal viremia (59,70).
An advantage of using mouse models to study congenital infection is the ability to use dams and sires of different IFN receptor genotypes to produce pregnancies with different IFN responsiveness on each side of the maternal-fetal interface, or among fetuses within a single pregnancy, as the placenta is a fetal-derived tissue and each fetus produces its own placenta. For example, crossing an Ifnar1 −/− dam and Ifnar1 +/− sire yields a pregnancy in which 50% of fetuses (and their associated placentas) are Ifnar1 −/− (lack type I IFN signaling) and 50% are Ifnar1 +/− (intact type I IFN signaling), within an Ifnar1 −/− dam.
Both type I and type III IFN signaling have an antiviral effect at the maternal-fetal interface and limit ZIKV transplacental transmission in mouse models. However, Ifnar1+/ − fetuses within Ifnar1 −/− dams succumb to ZIKV infection, indicating that fetal and placental type I IFN signaling are not sufficient to restrict congenital ZIKV infection (49,70,118). Somewhat counterintuitively, Ifnar1 −/− fetuses within Ifnar1 −/− dams exhibited less ZIKV-induced pathology than their Ifnar1+/ − littermates, suggesting a detrimental effect of type I IFN signaling on the developing placenta and fetus, and underscoring the delicate balance required of maternal antiviral immunity in the context of congenital infection (118). Uncontrolled viral replication and dysregulated cytokine signaling in Ifnar1 −/− mice result in high serum concentrations of type I IFN (60,86), which could contribute to severe infection-induced pathology in Ifnar1+/ − fetuses within Ifnar1 −/− dams. Moreover, type I IFN-mediated pathology is not a ZIKV-specific effect, as fetal pathology in Ifnar1+/ − fetuses can be induced by poly(I:C) (118). Other flaviviruses are able to produce congenital infection and fetal pathology in mice (52,89), so type I IFN-mediated fetal pathology may be a general effect of infection and morbidity in pregnant mice. Conversely, fetal and placental type I IFN signaling may reduce the severity of maternal viral infection (93). Type I IFN induction also is associated with preterm birth in mice following lymphocytic choriomeningitis virus or L. monocytogenes infection, as well as treatment with poly(I:C) or LPS (17). One mechanism for IFN-induced placental pathology could include ISGs that interfere with placental physiology. For example, IFITM proteins inhibit viral infection by interfering with membrane fusion (101), but IFITM expression in the placenta can disrupt cell fusion required for syncytiotrophoblast formation (15,120). Accordingly, mice lacking IFITMs are protected from IFN-induced pregnancy pathology (15).
Although Ifnar1 −/− mice have worsened disease outcomes in the context of viral infection, Ifnar1 −/− mice have reduced bacterial burdens following L. monocytogenes or C. muridarum infection (3,13,76). Accordingly, L. monocytogenes infection is controlled more effectively in pregnant Ifnar1 −/− dams compared to wild-type dams, including reduced bacterial burdens in the placenta and spleen as well as reduced fetal reabsorption rates (3).
NHP Models of Congenital ZIKV Infection
NHPs are a more physiologically relevant animal model for studying congenital ZIKV infections as they have singleton pregnancies, a hemochorial discoid placenta, and a long gestation (23 weeks in rhesus macaques). Pregnant rhesus macaques develop prolonged ZIKV viremia compared to nonpregnant macaques (30,65,77), consistent with prolonged viremia observed in ZIKV-infected pregnant women (29,83). Congenital ZIKV infection in rhesus macaques produces disease outcomes corresponding to those observed in humans, including fetal infection, placental pathology, neuropathology, ocular disease, and fetal loss (30,45,65,66,72,77). Rhesus macaques have become the most common NHP model of congenital ZIKV infection, although studies in pregnant pigtail macaques, olive baboons, and marmosets also observed restricted fetal brain growth, viral neuroinvasion, and neuroinflammation (1,2,42,102).
ZIKV infection induces a robust systemic adaptive immune response in both pregnant and nonpregnant NHP characterized by IFN-γ induction, leukocyte expansion, and ZIKV-specific IgG and IgM antibodies (30,45,46,62). During acute ZIKV infection in nonpregnant NHP, peripheral blood mononuclear cells (PBMC) exhibit rapid upregulation of type I and type II IFN, as well as ISGs (e.g., OASL, OAS2, IFIT1, MX1, MX2, and TRIM5) and proinflammatory cytokines and chemokines (e.g., TNFA, IL1, IL18, CCL2, and CCL20) (4). PBMC type I and type III IFN transcript levels correlate with the level of ZIKV viremia (4). ZIKV infection at the maternal-fetal interface also results in robust immune responses in the maternal decidua and in fetal tissues. Following infection, levels of IFN-γ and other proinflammatory cytokines (e.g., IL-2, IL-12, MIF, IL-1B, IL-2, IL-12, and IL-1RA) increased in the fetal blood (45).
During normal pregnancy, immunity in the decidua is tightly regulated to allow trophoblast invasion into decidual tissue and to prevent rejection of the semiallogeneic placenta. Once placentation is complete (the end of the first trimester, ∼3 weeks in rhesus macaques) (25), an anti-inflammatory immune milieu is maintained in the maternal decidua. Decidual leukocytes are dominated by a unique subset of NK cells with reduced cytotoxic potential, as well as Treg cells and M2-like macrophages (121). In ZIKV-infected pregnant rhesus macaques, inflammatory leukocytes, including nonclassical monocyte subsets and CD4+ T cells, were increased in the decidua and placental villous trees compared to uninfected controls late in gestation (135 days) (45). Although classical MHC molecules are not expressed on trophoblasts, Treg and Th2-type immune responses are necessary to prevent rejection of the fetus. Induction of IFN-γ and a proinflammatory Th1-type immune response at the maternal-fetal interface, for example resulting from viral infection, may contribute to placental damage and fetal demise during congenital ZIKV infection.
Th1-type immune responses also may be induced following ZIKV infection in mice. A subset of Th1-type T cells generally is excluded from the decidua during pregnancy due to epigenetic silencing of Cxcl9, Cxcl10, and Ccl5 (121). However, Cxcl9 and Cxcl10 were induced in the maternal decidua of ZIKV-infected mice, further suggesting that ZIKV infection disrupts the immune balance at the maternal-fetal interface (14).
Ex Vivo and Cell Culture Models of Placental Infection
ZIKV infection and IFN responses and production have been modeled using primary placental and decidual tissues cultured ex vivo. Explants from human mid-gestation and term placentas cultured ex vivo constitutively secrete type III IFN (10,18), which is among the mechanisms that render syncytiotrophoblasts refractory to viral infection (11,26). Following ZIKV infection, mid-gestation decidual explants produced type I and type III IFN (112) and placental macrophages produced IFN-α (but not IFN-β or IFN-λ) (92), suggesting additional sources of type I and III IFNs at the maternal-fetal interface. As maternally derived decidual cells respond to type I and type III IFNs, placenta-derived IFNs could induce an antiviral state on both sides of the maternal-fetal interface (18,49). Since type III IFN induces a less potent antiviral response than type I IFN, type III IFN signaling at the maternal-fetal interface may produce an antiviral state without the immune pathology induced by type I IFN (61). Accordingly, IFN-β induced pathological morphological changes (syncytial knots and sprouts) in human mid-gestation chorionic villous explants, whereas IFN-λ3 caused no pathology (118).
Dysregulated IFN Signaling in Autoimmunity and Adverse Pregnancy Outcomes
Women with dysregulated type I IFN signaling (sustained IFN production or impaired receptor downregulation) exhibit poor pregnancy outcomes. These include preeclampsia as well as neurodevelopmental defects similar to those induced by congenital infection, altogether consistent with a role for dysregulated type I IFN responses in placental damage (7,21,69,99). Elevated type I IFN levels during pregnancy may be pathogenic, since diseases involving increased production of type I IFN are associated with miscarriage (21,73). This association is most apparent in patients with systemic lupus erythematosus, a rheumatologic disease characterized by activation of the type I IFN response (9). Transcriptomic analysis of PBMCs from pregnant lupus patients revealed that preeclampsia and other fetal complications were associated with high ISG expression, in contrast to the downregulation of type I IFN responses observed during healthy pregnancy and in uncomplicated lupus pregnancy (47). A subset of patients with lupus develop antiphospholipid antibody syndrome, a disease that is associated with miscarriage, and in which patients produce antibodies that prolong clotting times in laboratory testing, but cause clots in vivo (32). Although lupus patients produce autoantibodies, the potentially pathogenic role of some autoantibodies remains uncertain. Nevertheless, one of the most compelling arguments for the pathogenicity of autoantibodies in lupus is the association between anti-SSA and anti-SSB autoantibodies and the development of neonatal lupus, a disease characterized by rash and other features of lupus in the first few months of life until maternal antibodies are cleared from the neonatal circulation (54). Neonatal lupus also can result in permanent damage to the conduction system of the developing heart, requiring pacemaker placement (39,71,105). Although the presence of maternal anti-SSA and SSB autoantibodies is strongly associated with risk of neonatal lupus, expression of high levels of type I IFN also may contribute to this disease entity (71,78). IFN-α may limit angiogenesis and affect the pathogenesis of preeclampsia in lupus pregnancy (7). In further support of the idea that IFN-α may contribute to neonatal lupus, IFN-α treatment in pregnancy was associated with IUGR and facial rash consistent with neonatal lupus in one case (34).
Rare monogenic diseases associated with increased production of type I IFN, known as interferonopathies, also provide insight into contributions of type I IFN to human disease pathogenesis. Some of the classic examples of monogenic interferonopathies include Aicardi-Goutieres Syndrome and STING-associated vasculopathy with onset in infancy (SAVI) (20,21,63). Both of these diseases are caused by mutations that upregulate the type I IFN response downstream of the TREX1-cGAS-STING pathway (24,116). Although these diseases are thought to be mediated by enhanced type I IFN production, this has not been definitively demonstrated in humans with monogenic interferonopathies. However, in an animal model of SAVI (heterozygous STING N153S knockin mice), disease is associated with lethality in pregnant dams and upregulation of ISGs in adult mice, although the mechanism of impaired survival during pregnancy was not determined (111). Autoimmunity in this model of interferonopathy also may reflect effects on adaptive immunity. For example, in addition to upregulation of the type I IFN response, STING N153S mice exhibit severe lung disease mediated primarily by T cells (64,114). Indeed, some features of the disease develop independent of IFNAR1, IRF3, and IRF7 (64), underscoring the complexity of defining the precise contributions of type I IFN in human studies, even when there is an associated type I IFN signature in patient cells.
Summary
Type I, II, and III IFNs serve important roles during pregnancy, both in controlling normal physiology at the maternal-fetal interface and in preventing transmission of maternal pathogens to the fetus. However, excessive or dysregulated IFN signaling may be pathogenic during pregnancies complicated by viral infection or systemic interferonopathies related to autoimmunity. Understanding the balance between protective and pathogenic effects of IFNs during pregnancy may lead to novel therapies to improve outcomes in complicated pregnancies associated with teratogenic infections or excessive production of IFN.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Research in the authors' laboratories is supported, in part, by R01 AI39512 (H.M.L.) and K08 AR070918 (J.J.M.), and R01 AI143982 (J.J.M.). R.L.C. is supported by T32 AI007419.