
Abstract
Zika virus (ZIKV) is a mosquito-transmitted flavivirus that caused a public health emergency in the Americas when an outbreak in Brazil became linked to congenital microcephaly. Understanding how ZIKV could evade the innate immune defenses of the mother, placenta, and fetus has become central to determining how the virus can traffic into the fetal brain. ZIKV, like other flaviviruses, evades host innate immune responses by leveraging viral proteins and other processes that occur during viral replication to allow spread to the placenta. Within the placenta, there are diverse cell types with coreceptors for ZIKV entry, creating an opportunity for the virus to establish a reservoir for replication and infect the fetus. The fetal brain is vulnerable to ZIKV, particularly during the first trimester, when it is beginning a dynamic process, to form highly complex and specialized regions orchestrated by neuroprogenitor cells. In this review, we provide a conceptual framework to understand the different routes for viral trafficking into the fetal brain and the eye, which are most likely to occur early and later in pregnancy. Based on the injury profile in human and nonhuman primates, ZIKV entry into the fetal brain likely occurs across both the blood/cerebrospinal fluid barrier in the choroid plexus and the blood/brain barrier. ZIKV can also enter the eye by trafficking across the blood/retinal barrier. Ultimately, the efficient escape of innate immune defenses by ZIKV is a key factor leading to viral infection. However, the host immune response against ZIKV can lead to injury and perturbations in developmental programs that drive cellular division, migration, and brain growth. The combined effect of innate immune evasion to facilitate viral propagation and the maternal/placental/fetal immune response to control the infection will determine the extent to which ZIKV can injure the fetal brain.
Introduction
The observation of an unexpected surge in congenital microcephaly occurring in tandem with the Zika virus (ZIKV) outbreak in Brazil in 2015–2016 led to declaration of a global public health emergency by the World Health Organization (1). ZIKV is a mosquito-transmitted flavivirus that typically leads to mild symptoms in infected adults, but may cause a spectrum of fetal brain injuries in pregnant women infected with ZIKV. The placenta is a permissive organ for viral replication, which can contribute to prolonged viremia in pregnant women (170). Unfortunately, the fetal brain and eye are especially vulnerable to ZIKV, where neural stem cells (NSCs) and neuroprogenitor cells are also permissive for infection and can facilitate further replication before cell death (49). As an emerging pathogen, the discrete molecular mechanisms conferring ZIKV pathogenesis and congenital disease outcomes remain incompletely resolved. In this review, we combine a focus on the molecular mechanisms contributing to innate immune evasion with the sequence of events likely to occur to facilitate ZIKV trafficking into the fetal brain and the eye. Understanding the mechanisms and biological events contributing to maternal/fetal transmission and viral trafficking into the brain is an important milestone toward developing antiviral therapeutics or vaccines for fetal protection.
How ZIKV Escapes the Host Immune Response
As with many related flaviviruses, ZIKV is transmitted to human hosts through the bite of an infected Aedes spp. mosquito (38,81,95,106). For most infected individuals, initial transmission is followed by an acute, rapid amplification of virus in peripheral tissues, with subsequent clearance via the host immune response. Unusually among the genus Flavivirus, ZIKV appears to be able to mediate a persistent infectious presence in the reproductive system and other sites for weeks or months following exposure (5,18,61,64,133,148). The balance between persistence and clearance of an infecting viral pathogen is governed by the efficiency of the host immune response, as well as the impact of viral-encoded countermeasures against antiviral immunity.
The host immune defense against ZIKV and related flaviviruses is broadly partitioned into two phases. The innate immune response comprises the first stage of pathogen recognition and relatively nonspecific antiviral defense initiation, and is essential for priming and shaping an efficient and specific adaptive immune response against infection (140,167). Innate immunity to ZIKV is triggered when viral RNA pathogen-associated molecular patterns (PAMP) are recognized by host cell retinoic acid inducible gene-I (RIG-I), one of the RIG-I-like receptors (RLRs) (34,58). This process leads to activation of latent transcription factors, including interferon regulatory factor (IRF) 3 and NF-κB, to induce the expression of innate immune genes (58,181). Innate immune gene products function as antiviral factors to restrict ZIKV replication and spread (157). IRF3 activation also leads to induction of type I and III interferon (IFN); IFN signaling through the Jak-STAT pathway then drives the induction of hundreds of IFN-stimulated genes mediating immune modulation and antiviral defense (98,157). However, ZIKV can block Jak-STAT signaling to evade host cell innate immune defenses (28,29,66,71,89). Inadequate programming of innate immunity can facilitate a disease state, in which ZIKV faces reduced barriers to dissemination and transmission. Compared with avirulent isolates, the most pathogenic flavivirus strains have been associated with a more efficient propensity to antagonize host innate immunity (83,105). Indeed, recent evidence has demonstrated that the same is true for ZIKV isolates, with differential innate immune activation and antagonism between African and Asian lineages (10,54,58,68,142,155,159,172,178).
A vast body of research has revealed that flaviviruses facilitate efficient replication and dissemination within an infected vertebrate host by direct antagonism and/or evasion of host innate and adaptive immunity via a plethora of discrete mechanisms. To provide a broad overview of these flavivirus immune antagonism and evasion phenotypes, we summarized evidence from decades of research in Table 1, dividing identified strategies into the following broad categories: (1) Viral proteins, (2) RNA elements, (3) Secreted viral particles, and (4) Replication cycle effects. It is becoming clear that ZIKV efficiently disrupts the host immune response using many mechanisms similar to other flaviviruses (Table 1). Considering this unfolding trend, as well as the conservation of flavivirus protein structure, genome structure, and replication strategy, we posit that summarizing evidence of immune escape described for related flaviviruses provides a roadmap for future ZIKV studies. We expect that data will emerge as research progresses, demonstrating ZIKV utilization of many of the strategies described in Table 1, with examples from each of the four antagonism/evasion categories.
Strategies of Flavivirus Antagonism and/or Evasion of the Vertebrate Immune Response, with Evidence for Zika Virus Involvement
Flaviviruses use a diverse set of strategies to inhibit the host immune response, which involve viral proteins, RNA elements, secreted viral particles, and replication cycle effects. The table categorizes known mechanisms of host immune evasion and signaling pathways targeted by specific flaviviruses (ZIKV shown in bold).
Ago2, protein argonaute-2; AP-1, activator protein 1; C, capsid protein; C4BP, C4 binding protein; C9, complement component 9; cGAS, cyclic GMP-AMP synthase; DENV, dengue virus; DRP1, dynamin-related protein 1; dsRNA, double-stranded RNA; DTMUV, duck Tembusu virus; E, envelope protein; elF2α, eukaryotic initiation factor 2 alpha; ER, endoplasmic reticulum; FMRP, fragile x mental retardation protein; G3BP1, GTPase-activating protein binding protein 1; HBV, hepatitis B virus; HO-1, heme oxygenase-1; IFIT1, interferon induced protein with tetratricopeptide repeats 1; IFN, interferon; IFNAR1, interferon alpha & beta receptor subunit 1; IGUV, Iguape virus; IKKɛ, ikappaB kinase epsilon; IL-6; interleukin 6; ISG, interferon-stimulated gene; ISGF3, interferon-stimulated gene factor 3; ISRE, interferon-sensitive response element; I2 PP2A, inhibitor-2 of protein phosphatase-2A; JEV, Japanese encephalitis virus; KFDV, Kyasanur forest disease virus; KPNA, karyopherin subunit; LGTV, Langat virus; MAC, membrane attack complex; MAM, mitochondria-associated ER membrane; MAVS, mitochondrial antiviral signaling protein; MDA5, melanoma differentiation-associated gene 5; MEF, murine embryonic fibroblast; MTase, methyltransferase; MVEV, Murray Valley encephalitis virus; MxA, myxovirus-resistance protein A; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NK, natural killer; NS, nonstructural protein; Pex19, peroxisomal biogenesis factor 19; pIRF3, phosphorylated interferon regulatory factor 3; PKR, protein kinase R; Poly(I:C), polyinosinic-polycytidylic acid; PP2A, protein phosphatase 2A; prM, premembrane protein; pSTAT1, phosphorylated signal transducer and activator of transcription 1; RdRp, RNA-dependent RNA-polymerase; RIG-I, retinoic acid inducible gene-I; RIP-1, receptor interacting protein kinase 1; RLR, RIG-I-like-receptor; RNAi, RNA interference; ROCV, Rocio virus; sfRNA, subgenomic flavivirus RNA; shRNA, small hairpin RNA; siRNA, small interfering RNA; SOCS1, suppressor of cytokine signaling 1; SPOV, Spondweni virus; STAT, signal transducer and activator of transcription; STING, stimulator of interferon genes; SVP, subviral particle; TAM, Tyro3, Axl, and Mer protein kinase family; TBEV, tick-borne encephalitis virus; TBK1, TANK-binding kinase 1; TIM, transmembrane immunoglobulin and mucin domain; TLR, toll-like receptor; TRAF6, tumor necrosis factor receptor-associated factor 6; TRIM25, tripartite motif containing 25; TYK2, tyrosine kinase 2; USUV, Usutu virus; UBR4, ubiquitin protein ligase E3 component N-recognin 4; WNV, West Nile virus; XBP1, X-box binding protein 1; YFV, yellow fever virus; YOKV, Yokose virus; ZIKV, Zika virus.
In fact, studies thus far show that ZIKV leverages every component of flavivirus replication to counter host immune responses. The viral proteins produced by ZIKV and related flaviviruses inhibit host immunity at several points (Table 1). Examples of strategies include the inhibition of C9 complement multimerization and deposition of the membrane attack complex on cells by NS1 (recombinant hexameric ZIKV NS1 when preincubated with normal human serum) (41); cleavage of human STING (stimulator of IFN genes) in vitro after overexpression of ZIKV NS2B/3 protease, which may inhibit cytoplasmic DNA-triggered innate responses (52); and viral antagonism of host type I IFN by NS5 when overexpressed by proteasomal degradation of human STAT2, thus attenuating the response to IFN [reviewed in (21)].
Specifically regarding PAMP recognition and IFN induction, the ZIKV proteins NS1, NS4A, NS4B, and NS5 have been demonstrated to play key roles in innate immune evasion (Table 1). Recombinant NS1 expression and the construction of ZIKV with mutations at NS1 amino acid residue 188 revealed this viral protein is able to bind to TANK-binding kinase 1 (TBK1) and inhibit RIG-I-dependent signaling (180). However, as flavivirus NS1 is solely localized to the endoplasmic reticulum lumen and is secreted from cells, questions remain as to how this viral protein can interact with TBK1, which is itself localized within the cytoplasm where it acts in antiviral signaling. Recombinant expression of ZIKV NS4A was able to block RLR-mediated induction of IFNβ, IRF3 activation, and induction of interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) promoter reporter constructs, while tagged NS4A was able to bind the RLR pathway signaling adaptor mitochondrial antiviral signaling protein (MAVS) in protein overexpression studies (88,112). ZIKV strains from Uganda and French Polynesia, as well as multiple strains of dengue virus (DENV), were shown to induce mitochondrial elongation within infected cells (33). This phenotype was recapitulated via overexpression of DENV NS4B alone and was attributed to inactivation of the mitochondrial fission-promoting factor dynamin-related protein1 (DRP1). DRP1 inactivation, leading to mitochondrial elongation, prevented the induction of types I and III IFN, and of IFIT1 in response to DENV infection, likely due to disruption of the mitochondrial-associated membrane, which acts as an RLR signaling platform (33). This mechanism of innate immune regulation is likely to be shared with ZIKV. Similar to NS4A, overexpression of ZIKV NS5 was also demonstrated to inhibit RLR-mediated promoter/reporter construct induction (72,88). This regulation was subsequently demonstrated to be due to NS5 interaction with TBK1, which reduces activating phosphorylation of this factor and prevents TBK1-TRAF6 complex formation (101).
Elements of the flavivirus RNA genome also contribute to host immune evasion, which has also been shown for ZIKV (Table 1). For flaviviruses in general, the 2′-O-methylated genomic RNA cap resembles an authentic host mRNA cap, and so evades IFIT1 recognition and abrogation of translation (32,47,85,168). In addition, cytoplasmic accumulation of ZIKV and other flavivirus 3′ UTR-derived subgenomic flavivirus RNA (sfRNA) has been demonstrated to antagonize immunity, with DENV sfRNA binding to TRIM25 and inhibiting RLR-induced antiviral signaling, among other activities (53,114). The particles secreted during flavivirus infection likewise counter host immunity (Table 1). One example includes phosphatidylserine incorporation into the virion envelope allowing engagement of TAM receptors such as Axl to induce suppressor of cytokine signaling (SOCS) 1 in infected cells and inhibit the type I IFN responses (35,119). Finally, perturbation of the cellular environment due to replication cycle effects assists flavivirus antagonism of host immunity via several described mechanisms (Table 1). Examples include MHC-I upregulation on the surface of infected cells, which evades NK cell-mediated lysis during coincubation in vitro (62); and stimulation of the ER unfolded protein response to inhibit type I IFN signaling (7,8,171). ZIKV, like many flaviviruses, utilizes elements of the RNA genome, secreted particles, and indirect effects of replication within host cells to evade the host immune response.
Although primarily identified and assessed in cells of nonreproductive origin, these diverse viral mechanisms to both evade and antagonize host antiviral innate immunity are thought to act in concert to facilitate efficient ZIKV replication and dissemination throughout the infected host, leading ultimately to the virus crossing tissue barriers to reach the developing fetus. Research using the related flavivirus West Nile virus (WNV) has revealed that whole-body knockout of both MAVS and the type I IFN receptor subunit IFNAR1 led to enhanced tissue tropism of infected mice, with increased WNV burden within a normally nonpermissive site (liver) and a collateral lack of virus-responsive gene induction (166). This outcome directly demonstrated that innate immunity is a major component of tissue protection, allowing inference that virus antagonism/evasion of the innate response permits access and spread to tropic tissue sites. Furthermore, a promising immunocompetent ZIKV mouse model has been recently developed that utilizes knockin (KI) of human STAT2 (65). Unlike human STAT2, mouse STAT2 is resistant to ZIKV NS5 binding and proteasomal degradation (66,88), and so pregnant wild-type (WT) mice are generally able to clear ZIKV infection and prevent virus dissemination to the fetus. Infection of pregnant STAT2 KI mice with a mouse-adapted Dakar strain of ZIKV led to virus crossing the placental barrier and infecting the fetal central nervous system, in stark contrast with WT mice infected in parallel (65). As the only difference between these mice was the species origin of STAT2, the NS5-STAT2 axis of ZIKV IFN antagonism was demonstrated as a crucial determining factor regulating viral spread to the developing fetus.
How ZIKV Traffics from the Mother into the Fetus
Understanding the path taken by ZIKV to reach the fetus and the probable tissues and cellular targets of infection is ideal for studying innate immune evasion in relevant cell types. Once ZIKV enters the maternal bloodstream, it is highly likely to encounter the placenta, which receives 500–700 mL of maternal blood every minute in the third trimester. There are many cell types at the maternal/fetal interface that express viral cofactors such as Axl, Tyro3, and/or TIM1, which can facilitate viral entry, replication, and eventual transmission to the fetus (170). ZIKV can replicate in a variety of primary cell types isolated from both the placental chorionic villi and the chorioamniotic membranes (e.g., cytotrophoblast cells, Hofbauer cells, amniotic epithelial cells, and umbilical cord endothelial cells) (55,79,139,170). Similarly, ZIKV can infect diverse cell types, including the proliferating cytotrophoblasts in cell columns; in contrast, syncytiotrophoblasts are resistant to infection, likely due to the actions of IFN-λ (78,169,170). Although the ontogeny of innate immune signaling pathways in the placenta is poorly understood, antiviral pathways are likely impaired in early pregnancy when rates of congenital microcephaly are highest (170). Overall, a number of cell types in different placental tissues are permissive for ZIKV infection and replication.
After breaching the placental “barrier” and entering the fetal circulation, ZIKV encounters specialized barrier systems regulating metabolite, gas, growth factor, and nutrient exchange between the vasculature and the developing nervous system (163). Barrier systems for the nervous system include the blood/cerebrospinal fluid (CSF) barrier within the choroid plexus regulating access to the interior brain and spinal cord surfaces, the blood/brain barrier (BBB) regulating access to the outer brain and spinal cord regions, and the blood/retinal barrier regulating access to the developing eye. Importantly, these barrier systems also normally protect the nervous system from external insults and immune cell entry, attack, and damage (163). Hence, the developmental timing of barrier integrity irrespective of immune activation would also be expected to impact the extent of ZIKV disease in the nervous system.
The Choroid Plexus: A Highway into the Fetal Brain
The choroid plexus is an early developing and disproportionately large vascular structure in the fetal brain that functions as a major “highway” for rapid and direct trafficking of gases, nutrients, and growth factors from the fetal blood into the CSF (Fig. 1). The choroid plexus is a complex structure and in vitro models are lacking that could recapitulate ZIKV trafficking into the fetal brain. Once ZIKV passes through the choroid plexus into the CSF, it has direct access to all internal brain and spinal cord surfaces before development of the surrounding perineural vascular plexus and subsequent blood vessel invasion (163). Unfortunately, the choroid plexus is highly vulnerable to pathogens and can be infected with DENV and chikungunya virus, as well as other RNA viruses (91). The blood/CSF barrier within the choroid plexus may be susceptible to ZIKV infection through exposure of multiple vulnerable cell types (Fig. 1A; e.g., endothelial cells and fetal macrophages). Alternatively, ZIKV may traffic across the choroid epithelium via transcytosis, which involves endocytosis of virus captured from the choroid plexus stroma, followed by transport across the cell and exocytosis into the CSF. Although choroid plexus infection by ZIKV has not yet been demonstrated, injury patterns in both humans and nonhuman primates focused around the lateral ventricles (which line the CSF) are consistent with this hypothesis (2). Once inside the CSF, ZIKV could gain access to all internal brain and spinal cord surfaces.

Early ZIKV highway through the BCB within the choroid plexus into the internal fetal brain. This illustration depicts the locations of the BCB in the choroid plexus of a developing human fetal brain. Note that the choroid plexus is a disproportionately large structure in the fetal brain and functions to generate CSF and transport metabolites, gases, growth factors, and nutrients into the brain. In this diagram, ZIKV traffics across the fenestrated capillary within the choroid plexus and then across the stroma where it infects a fetal macrophage. Then, ZIKV can then enter the CSF by directly trafficking across the choroid plexus epithelium. BCB, blood/CSF barrier, CSF, cerebrospinal fluid; RBC, red blood cell; ZIKV, Zika virus.
Many in vitro and organoid models have demonstrated the vulnerability of NSCs to ZIKV infection (46,97,135); NSCs are exposed to the CSF and vulnerable to ZIKV once across the blood/choroid plexus barrier. During early fetal development, cortical NSCs, also called radial glia, and intermediate progenitors (IPs, specialized neurogenic daughter cells) are located in a proliferative niche along the ventricular surface, directly juxtaposed to the CSF (Fig. 2A) (27,69,132). Hence, trafficking across the blood/CSF barrier early in gestation allows ZIKV to infect and kill fetal NSCs and IPs along ventricular surfaces, resulting in severe brain injury and even fetal demise (Fig. 2A). At later stages, most NSCs and IPs lose contact with the CSF, migrate away from inner zones into transient outer neurogenic niches in the overlying cortex, making neurons and glia before eventually disappearing (Fig. 2B) (69,132). However, many NSCs and IPs remain in this periventricular region, continuing to proliferate, making different neuron and glial subtypes important for late brain development and generating the ependymal epithelia, a new layer separating CSF from internal brain surfaces and important for CSF flow (Fig. 2B) (27).
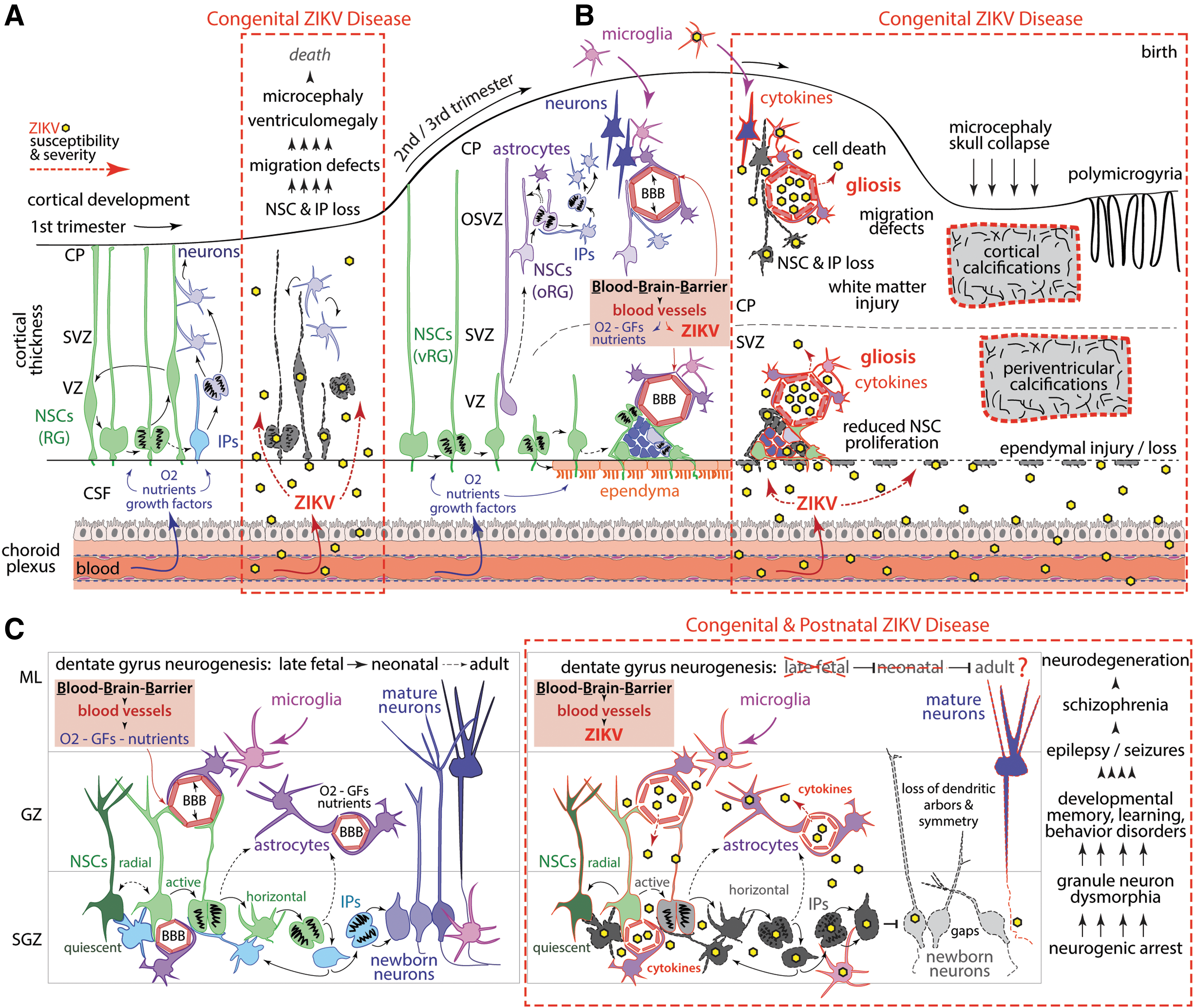
ZIKV trafficking into the fetal cortex and hippocampus. This illustration depicts normal development and the complex infection cascade (red dashed lines) within the fetal cortex
Several observations implicate ZIKV trafficking across the blood/CSF barrier within the choroid plexus (Figs. 1, 2). A consistent and striking periventricular injury pattern was demonstrated in nonhuman primate models of congenital ZIKV infection (1,39,67,116). Brain imaging revealed periventricular cortical T2-hyperintese foci coinciding with decelerated fetal brain growth (1). Neuropathology identified ependymal injury and loss along the posterior lateral ventricles and glial fibrillary acidic protein-immunoreactive periventricular white matter gliosis (1). In other ZIKV studies, similar periventricular neuropathology and pathology within the spinal cord were also observed (39,67,116). Since the ependymal cell layer in the cortex normally arises rather late during fetal human development and continues to mature long after birth (40), its protective function would be limited. Virus trafficking across an incompletely developed ependymal cell layer would then come into contact with NSCs in the subventricular zone (Fig. 2A). Indeed, in a nonhuman primate model, a maternal ZIKV infection was associated with a reduction in Ki67+ cells (reduced NSC proliferation) along the subventricular zone, indicating compromised neurogenic output (Fig. 2A) (1,27,39). Depending on ependymal developmental state (40), a ZIKV-mediated ependymal injury could further impair CSF transport and outflow through the dura, either of which might contribute to ventriculomegaly (and rarely hydrocephalus), a finding in both nonhuman primate ZIKV studies and human studies (115).
The BBB: A Second Entry Point for ZIKV into the Fetal Brain
Other ZIKV neuropathologic findings of cortical/subcortical calcifications indicate that cells in the outer brain regions without access to CSF are vulnerable to ZIKV, which implicates viral trafficking across the BBB. The mature BBB is composed of specialized endothelial cells connected by tight junctions, pericytes, and astrocyte processes (glial limitans), together regulating metabolite, gas, growth factor, and nutrient exchange to nourish cells in outer brain regions and prevent pathogen entry into the brain (134). BBB development begins as endothelial cells from the surrounding perineural vascular plexus migrate into stereotypical neuroectodermal regions governed by local NSC and neuron signaling interactions (14,19,25,134,147); these endothelial cells serve to direct nervous system-specific phenotypes and functions distinct from other vasculatures. Hence, BBB development is closely timed to normal developmental trajectories of the nervous system. The gestational timing of the BBB maturation, specifically in regard to pathogen defense, is unknown. Furthermore, both the blood/CSF barrier and BBB may become disrupted by inflammatory cytokines increasing the permeability of both barriers (91,134,163). Endothelial cells are also known to be vulnerable to ZIKV infection due to their expression of Axl (102).
Posterior cortical/subcortical calcifications and gliosis, and late migration defects (polymicrogyria), suggest ZIKV also traffics through the BBB to access outer brain cells at later stages of fetal development (Fig. 2B), contributing to motor, vision, auditory, arthrogryposis, and dystonic disorders (146). Although vascular defects alone can contribute to secondary calcifications and polymicrogyria, endothelial cells seem to readily transport ZIKV without disruption and serve as a potential ZIKV reservoir (129,137). Once transported into the outer brain parenchyma through the BBB, ZIKV would have ready access to outer NSCs, IPs, neurons, as well as astrocytes and microglia, later in gestation (Fig. 2B). Depending on ZIKV strain and tropism, infection in outer brain cells leads to loss of progenitors and secondary neuroinflammation accompanied by further infiltration of activated immune cells and cytokine exposure, which itself suppresses neurogenesis. In addition, loss of outer NSC-derived angiogenic signaling would further exacerbate vascular defects.
Additional supporting evidence for BBB-mediated ZIKV entry into outer brain regions is observed in the hippocampus, a key brain region that mediates flexible learning, memory, and anxiety/emotion regulation (9,63). A specialized subregion of the hippocampus called the dentate gyrus maintains a population of NSCs that establish a neurogenic niche, which is maintained through birth and childhood (Fig. 2C) (20,26,63,123,160,161). ZIKV-exposed fetal nonhuman primates exhibited a striking loss of neurogenic output in the dentate gyrus subgranular zone (1). Immunohistochemistry revealed that while dentate gyrus NSCs (Sox2+ cells) were still present, NSCs were highly disorganized due to ZIKV exposure, and IPs (Tbr2+/EOMES+ cells) were significantly reduced indicating neurogenic arrest (1). Remaining newborn neurons (doublecortin+ cells) were dysmorphic, and the entire granule cell layer was disrupted (1). This tissue phenotype links with epilepsy and seizures, a common occurrence in children living with congenital ZIKV disease (146). Another report using a different ZIKV inoculate, timing, and species (baboon) also found a reduction in dentate gyrus Sox2+ NSCs (67). Moreover, a postnatal ZIKV infection in young rhesus macaques also resulted in progressive hippocampal volume reduction and connectivity associated with behavioral changes (117). These observations further support BBB ZIKV trafficking, since this neurogenic niche is not in direct contact with the CSF.
Blood/Retinal Barrier: Viral Access to the Eye
The most visible protective barrier to the nervous system is the blood/retinal barrier, which functions to supply and exchange metabolites, gases, growth factors, and nutrients to the external and internal eye tissues (163). Since the eye and retina are vulnerable to ZIKV injury, and conjunctivitis and uveitis (dilated, inflamed eye capillaries) are one of the most common signs of ZIKV infection in children and adults (96,115,158,163), an understanding of eye development and how the timing of ZIKV-exposure might generate retinal pathology is important to predict the potential impact to the visual system. Recent studies reveal that the human retina develops through key stages in a central to peripheral manner (76). ZIKV exposure during early optic cup formation would allow access to all anterior chamber and neural retinal stem cells, leading to possible eye loss and/or microphthalmia. Importantly, case series in human newborns with congenital Zika syndrome describe eye findings that are consistent with this profile of injury (50,115). Early neurogenic stages are dominated by central NSCs and ganglion cells, which are responsible for growing axons through the optic nerve to connect with central visual pathways; the peripheral retina is also packed with NSCs, which are vulnerable to ZIKV infection (Fig. 3A). Since the outer blood/retinal barrier is primarily responsible for the initial vascular supply to the eye, ZIKV exposure during the first trimester would grant access to the retinal pigment epithelium, leading to NSC death in the retina, epithelial mottling and lesion, chorioretinal scarring, and macular atrophy (Fig. 3A) (115,158).
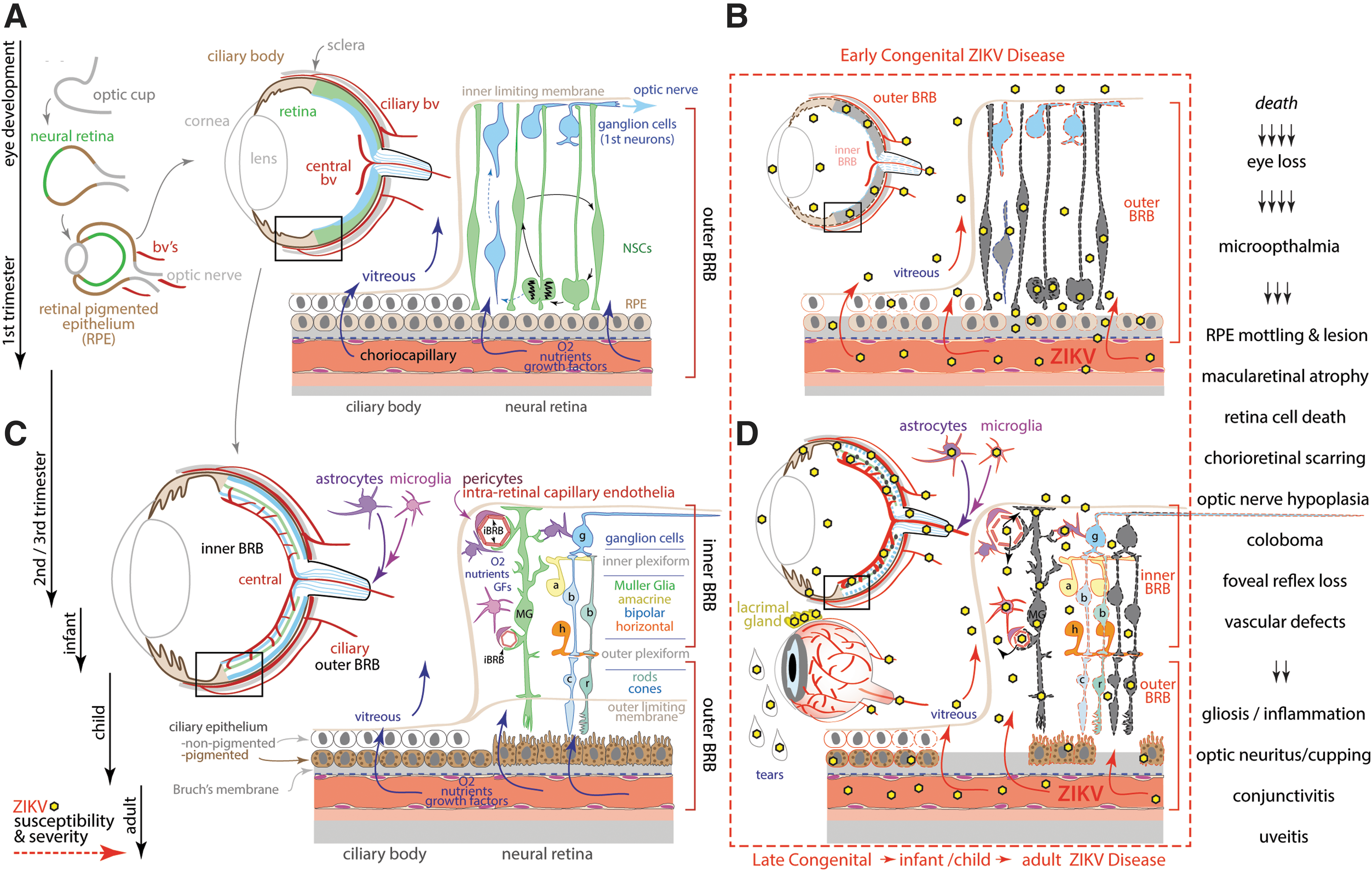
ZIKV trafficking into the eye and retina.
Although the eye becomes less accessible to overt teratogenic damage over time as the blood/retinal barrier matures, it is still susceptible to insults leading to impaired vision and even progressive blindness during later fetal stages and into adulthood; ocular injuries associated with ZIKV infection described in the literature include optic nerve hypoplasia, coloboma, foveal reflex loss, vascular defects, and especially inflammation at later fetal stages mediating gliosis and optic nerve cupping/neuritis (115,158). Experimental injection of ZIKV into WT adult mice is associated with ZIKV infection of retinal cells (156,189), largely infecting postmitotic Müller glial cells that differentiate from the last retinal NSC (130). Since Müller glial cells regulate both the inner and outer blood/retinal barrier and provide key trophic support for all other retinal neurons (Fig. 3B), Muller glial cell loss indirectly impacts remaining cells, leading to progressive retinopathy. Furthermore, immune-compromised adult mice exhibited widespread ZIKV infection and retinal cell death (156). Finally, a visibly dilated blood/retinal barrier in the eyes of infants, children, and adults (uveitis) is strongly indicative that ZIKV trafficking occurred into the eye and brain across the blood/retinal barrier, BBB, and blood/CSF barrier.
Conclusion
ZIKV continues to emerge as a public health threat to pregnant women in large parts of the Americas, Asia, and Africa. The pathogenesis of congenital microcephaly associated with ZIKV infection during pregnancy represents a complex series of events beginning with viral infection, subversion of host immunity, and trafficking across multiple placental and fetal tissue barriers. Evasion of host immunity is a fundamental event that facilitates ZIKV infection, viral propagation, and spread to the fetus. ZIKV shares many mechanisms with other flaviviruses to evade antiviral immunity that involve viral proteins, genomic RNA elements, secreted particles, and processes occurring in the course of viral replication. There is a great need to understand the mechanisms of how ZIKV infection and host immune evasion occur in the context of relevant tissues and cell types encountered during viral trafficking into the fetal brain and eye. Once ZIKV has entered the fetal circulation, it is likely to cross several different barriers to induce fetal injury: the blood/CSF barrier (choroid plexus), BBB, and the blood/retinal barrier. The gestational timing of the maturation of innate immune antiviral pathways within fetal tissues and cell types is unknown and warrants focused research. Overall, fetal immunity is likely highly impaired in the first trimester when the risk for congenital ZIKV infection and microcephaly outcome is the greatest. Finally, insights into the pathogenesis of the congenital Zika syndrome is most likely to occur through a linked consideration of innate immune evasion within the key cell types of the placenta and barriers to infection of the fetal brain and eye.
Footnotes
Acknowledgment
We acknowledge and thank Jan Hamanishi for her assistance in preparing the figures.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other funders.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Institutes of Health, grant numbers R01AI133976 (L.R and K.M.A.W), AI144938 (K.M.A.W), OD010425 (K.M.A.W. and M.G.), AI43265 (K.M.A.W. and M.G.), AI083019 (M.G.), AI104002 (M.G.), and AI145296 (M.G.). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.