
Abstract
Males and females respond to pathogens differently and exhibit significantly different frequencies of autoimmune disease. For example, vaccinated adult females control influenza virus better than males, but females suffer systemic lupus erythematosus at a 9:1 frequency compared to males. Numerous explanations have been offered for these sex differences, but most have involved indirect mechanisms by which estrogen, a nuclear hormone, modifies cell barriers or immunity. In search of a direct mechanism, we examined the binding of estrogen receptor α (ERα), a class I nuclear hormone receptor, to the immunoglobulin heavy chain locus. Here, we show that in purified murine B cells, ERα and RNA polymerase II (RNA Pol II) exhibit extraordinarily similar DNA binding patterns. We further demonstrate that ERα preferentially binds adenosine–cytidine (AC)-repeats in the immunoglobulin heavy chain locus when supplemental estrogen is added to purified, lipopolysaccharide-activated B cells. Based on these and previous data, we hypothesize that (i) estrogen guides the binding of ERα and its RNA Pol II partner within the locus, which in turn instructs sterile transcription and class switch recombination (CSR), (ii) ERα binding to AC-repeats modifies the DNA architecture and loops associated with CSR, and (iii) by these mechanisms, estrogen instructs antibody expression. By targeting ERα-DNA interactions in the immunoglobulin heavy chain locus, clinicians may ultimately enhance antibody responses in the context of infectious diseases and reduce antibody responses in the context of allergic or autoimmune reactions.
Introduction
Different immune responses in females and males
Females generally express higher levels of serum immunoglobulins compared to males, with a tendency to produce more IgG1 and IgG2 (47,68,88). As a consequence of increased antibody production, females may have an advantage over males in the clearance of certain pathogens. For example, the immune response to influenza virus is better in vaccinated adult females compared to males. Furthermore, males often experience worse disease symptoms after an influenza virus infection compared to females (47,58,90). But enhanced antibody responses in females may come at a cost. Estrogen induces anti-self antibodies and females have extremely high frequencies of the autoimmune disease systemic lupus erythematosus (“lupus”) compared to males (14,16,75,94). During pregnancy, when estrogen may rise to levels of >6,000 pg/mL [compared to levels of <100 pg/mL in males (36,44)], lupus can be life threatening (14,16,18,65,71,75,78).
Differences between females and males in immune responsiveness depend on the environment and target antigens. In vitamin A deficient (VAD) C57BL/6 mice, the female:male differences in IgG2b expression are reversed compared to non-VAD mice (47). Moreover, in the context of a pneumococcus vaccination and infection, C57BL/6 male mice are better protected than females (47).
B cells and class switch recombination
The expression of IgG (including IgG3, IgG1, IgG2b, and IgG2c in C57BL/6 mice), IgE and IgA depends on class switch recombination (CSR), which occurs in B cells following antigen or mitogen stimulation to associate V-D-J gene segments with a constant (C) region gene segment (Cγ, Cɛ, Cα) downstream of Cμ. CSR involves cleavage of DNA in switch (S) regions positioned upstream of C gene segments (e.g., Sμ and Sγ1) and the ligation of two different S regions to delete intervening sequences (86). During CSR, DNA loops are observed that juxtapose promoters, S regions, and enhancers (87).
One of the first steps in CSR is the initiation of sterile germline transcription by RNA Polymerase II (RNA Pol II, spliced transcripts may be integral to the CSR mechanism) (50,66). R loops form (comprising an RNA–DNA hybrid and a single strand of DNA) and stalled RNA Pol II recruits activation induced deaminase (AID) (12,13,50,73). Subsequent steps can include conversion of dC to dU by AID, excision of dU bases by uracil DNA glycosylase (UNG), DNA cleavage by abasic endonucleases, and ligation of cleaved DNA either within or between S regions by nonhomologous end-joining machinery (47).
Multiple enhancers are present in the murine immunoglobulin heavy chain locus, including the Eμ enhancer upstream of Cμ and enhancers within the 3′ regulatory region (3′RR) downstream of Cα (7,56,89). The 3′RR is rich in DNase1 hypersensitive sites (HS) and is required both for CSR and somatic hypermutation (SHM). In mice, a span of 40 kb covers two discrete regions. The upstream segment (∼28 kb) includes four HS sites, HS3A, HS1,2, HS3B, and HS4. The downstream segment (∼12 kb) includes HS5-8, an insulator region (7). In humans, the heavy chain locus is configured differently than in mice. There are two distinct 3′RR regions, one downstream of Cα1, and one downstream of Cα2. As in mice, these 3′RR regions influence CSR and antibody expression. Notably, a human polymorphism that involves an HS1,2 duplication is associated with a significant increase in frequencies of lupus (29,32).
Looping and CSR are influenced by enhanceosomes, the protein complexes associated with enhancers (53). Many proteins are associated with the HS1–4 complex, including Mediator (92), the CCATT enhancer binding protein (CEBP), the octamer binding protein, Pax5/BSAP, and NFκB family members (7). In contrast, CCCTC-binding factor (CTCF) and the subunits of cohesin (SMC1, SMC3) (93) are preferentially bound farther downstream in the insulator region (6). Enhanceosomes containing CEBP, CTCF, and/or cohesin can independently support DNA loop formation. Throughout the genome, signature cooperative protein sets are observed (e.g., STAT5A-CEBPβ-PML or CTCF-RAD21-SMC3 trios [9,95,103,104]).
B cell activation under variable conditions will alter enhanceosome composition, both within and between HS regions (7). Knock-out (KO) mutations have revealed the complex influences of different HS sites on CSR and antibody expression. Deletion of HS3B and HS4 reduced CSR to all isotypes except IgG1 (15) while deletion of the entire upstream (∼28 kb) 3′RR limited CSR to all isotypes and prevented SHM (6,7,82).
Estrogen, the estrogen receptor, and the immunoglobulin heavy chain locus
Estrogen functions both within and outside of the nucleus, but is best known for its binding to the class I nuclear hormone receptor estrogen receptor α (ERα), a transcription factor that binds DNA to regulate gene transcription (30,57,99 –101). ERα often binds estrogen response elements (EREs, GGTCAnnnTGACC) (28,30,57,63), but ERα–DNA interactions can occur in the absence of an ERE and can be assisted by interactions with other factors including NFκB, AP-1, and SP1 (25,28,60,67,70,76,84). ERα may regulate gene transcription by direct binding to a promoter, although the ERα–DNA interactions responsible for gene regulation are often far more complex. For example, estrogen regulation of the GREB1 gene involves recruitment of ERα and RNA Pol II to three different ERE within 20 kb of upstream flanking sequences, chromatin loop formation, and juxtaposition of EREs with the transcriptional start site (21,91).
Because ERα will influence the functions of virtually every mammalian cell, there are many explanations for female/male differences in influenza virus-specific responses and autoimmune disease. As one example, estrogen may regulate innate cells that in turn regulate B cell functions (37,52). In addition, estrogen can upregulate AID, an enzyme integral to the initiation of CSR (61,69).
We previously hypothesized that ERα might also influence antibody expression by direct binding to the immunoglobulin heavy chain locus, and therefore queried the locus for ERE. In so doing, we discovered hotspots of response elements, both for type I and type II nuclear hormone receptors. These included ERE and retinoic acid response elements [two half-sites, PuG(G/T)TCA, often separated by a short spacer] (42). We then used chromatin immunoprecipitation (ChIP)-seq analyses to confirm that ERα was bound to DNA and found peak binding within enhancers known to influence CSR (46,47). Moreover, when ERE sequences were removed from enhancers in the heavy chain locus using clustered regularly interspaced short palindromic repeats (CRISPR)- CRISPR-associated protein-9 nuclease (Cas9) KO strategies in CH12F3.5B1 cells, the switch in isotype from IgM to IgA expression was inhibited (79).
Here, we examine additional features of ERα binding in the immunoglobulin heavy chain locus to dissect estrogen's influence on CSR and gene expression. We find that ERα and RNA Pol II binding patterns are strikingly similar in Eμ, Sμ, and the 3′RR in purified B cells, supporting our previous finding that supplemental estrogen in purified B cell cultures drives synchronous shifts in ERα and RNA Pol II binding within the locus (47). We also find a propensity for ERα binding to adenosine–cytidine (AC)-rich sequence repeats in the 3′RR of estrogen-supplemented B cell cultures. Results support our hypothesis that estrogen instructs the composition of enhanceosomes and assists DNA loop formation, explaining at least in part why males and females exhibit different antibody expression patterns and are variably susceptible to pathogens, allergies, and autoimmune disease.
Materials and Methods
ChIP-Seq libraries
The ChIP-seq library from lipopolysaccharide (LPS)-stimulated purified B cells has been described previously (46,47). Briefly, B cells were purified from the spleens of C57BL/6J mice by negative selection with anti-CD43 and anti-CD11b microbeads (Miltenyi Biotec) using a MACS LD Column (Miltenyi Biotec). Purified B cells were cultured in RPMI medium (Life Technologies) containing 10% fetal bovine serum, 2 mM
Harvested cells were treated with 2 mM disuccinimidyl glutarate (ProteoChem) in Dulbecco's phosphate buffered saline (DPBS) (Lonza) with proteinase inhibitors (PIs) phenylmethylsulfonyl fluoride (PMSF) (Sigma), Pepstatin A (Sigma), and Leupeptin (Sigma) and then washed and fixed in DPBS plus PIs and 1% paraformaldehyde (Sigma, Thermo Scientific) for 5 min with rotation at room temperature. The reaction was quenched with glycine (200 mM final concentration) and rotation for an additional 5 min. The cell pellet was washed with DPBS plus PIs and lysed in Covaris lysing buffer + PIs on ice for 10 min. Nuclei were centrifuged at 1500–1700 g for 5 min and washed 2 × with Covaris wash buffer and then 2 × with shearing buffer with PIs. The pellet was resuspended in Covaris shearing buffer plus PIs at a concentration of 1 mL per initial 2 × 107 cells and sheared in the Covaris E210 or E220 in Covaris MilliTubes with 200 cycles/burst, 20 W for 25–30 min. Sheared chromatin was diluted with Covaris ChIP dilution buffer and immunoprecipitated with anti-ERα antibody (Abcam; Cat#32063, monoclonal E115) or with anti-RNA Polymerase antibody (Active Motif Cat#61081) in combination with anti-mouse IgG bridging antibody (Active Motif Cat#102302) and Protein A/G magnetic beads. DNA was isolated from beads, purified, and quantified using the Quant-iT PicoGreen assay (Life Technologies) Qubit dsDNA HS Assay Kit (ThermoFisher Scientific) or SpectraMax Quant AccuBlue Pico dsDNA assay kit (Molecular Devices).
For the ERα studies with LPS or LPS + E cultured cells, libraries were prepared from DNA using the NEBNext ChIP-Seq Library Prep Reagent Set for Illumina with NEBNext Q5 Hot Start HiFi PCR Master Mix according to the manufacturer's instructions (New England Biolabs, Ipswich, MA) with a modification: a second 1:1 Ampure cleanup was added after adaptor ligation. For RNA Pol II studies with LPS-cultured cells, libraries were prepared from ∼1 to 10 ng of purified DNA using the KAPA HyperPrep Library Preparation Kit (Roche). Fifty cycle single-end or paired-end sequencing was performed on an Illumlina HiSeq 2000 or 2500, NovaSeq 6000, or NextSeq 550 instrument.
For ChIP-Seq data analysis, we followed the guideline of ENCODE for quality control (54). Details and codes have been previously described (19,54,102). The bigwig tracks were normalized to 15M uniquely mapped reads.
ENCODE ChIP-seq libraries were produced with purified C57BL/6J splenic B cells, negatively selected for CD43+ and CD11b+ cells. Bigwig tracks were downloaded from the ENCODE portal with the following identifiers: SERIES ENCSR902FHL, ENCSR000CBC, ENCSR000CBD, ENCSR000CBE, ENCSR000CBY, ENCSR000CBZ, ENCSR000CDJ, ENCSR000CFT, and ENCSR000CGA (17,19). Libraries were evaluated using Integrative Genomics Viewer (IGV) software.
Results
ERα and RNA Pol II co-bind the immunoglobulin heavy chain locus
We previously described ERα binding patterns in the immunoglobulin heavy chain locus of purified, LPS-activated C57BL/6 splenic B cells (46,47,79). Because RNA Pol II recruitment has been previously described as an integral step in estrogen-induced gene regulation (21,101), we queried the relationships between ERα and RNA Pol II binding within the immunoglobulin heavy chain locus. To this end, we aligned our ERα ChIP-seq data with RNA Pol II ChIP-seq data from ENCODE (Library ENCSR000CBZ, Target POLR2A). Both libraries originated from purified (negatively selected for CD43+ and CD11b+ cells), C57BL/6 splenic B cells, in one case collected after LPS stimulation. As shown in Figure 1, the alignment revealed strikingly similar binding patterns between ERα and RNA Pol II; each protein bound Sμ and 3′RR HS sites within the immunoglobulin heavy chain locus.
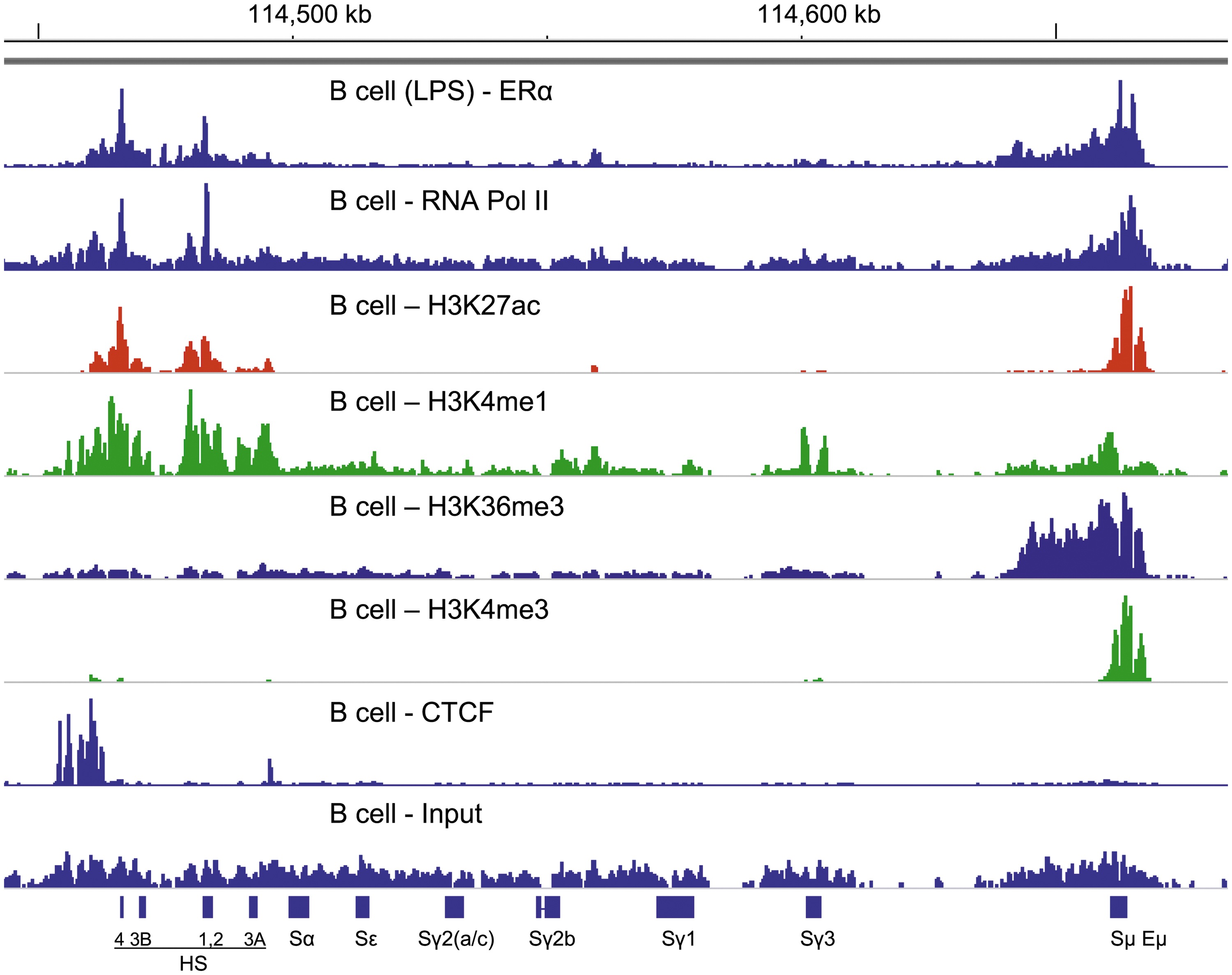
ERα binding and RNA Pol II binding patterns match. ChIP-seq libraries are aligned using IGV software (mm9, chromosome 12). The locations of switch regions and 3′RR enhancers are indicated. The ChIP-seq library with LPS-stimulated, purified splenic B cells from C57BL/6 mice was described previously (46,47,79) (range 0–138). Data were normalized against 15M uniquely mapped reads (102). Additional ChIP-seq libraries were from ENCODE. These used unstimulated, purified splenic B cells from C57BL/6 mice. ENCODE ChIP-seq libraries examined RNA Pol II (range 0–2.74), H3K27ac (range 0–37), H3K4me1 (range 0–4.41), H3K36me3 (range 0–3.38), H3K4me3 (range 0–31), and CTCF (range 0–18). For input, the range was 0–2.23. Patterns were most similar between ERα and RNA Pol II binding. ER, estrogen receptor; RNA Pol II, RNA polymerase II; CTCF, CCCTC binding factor; HS, (DNase I) hypersensitive site; ChIP, chromatin immunoprecipitation; LPS, lipopolysaccharide; RR, regulatory region; IGV, Integrative Genomics Viewer.
We aligned additional ENCODE ChIP-seq data (ENCSR000CBC, ENCSR000CBD, ENCSR000CBE, ENCSR000CBY, ENCSR000CDJ, ENCSR000CFT, and ENCSR000CGA) to examine positions of histone modifications and CTCF binding in the immunoglobulin heavy chain locus of purified B cells. We found that H3K27ac and H3K4me1 binding patterns were similar to ERα and RNA Pol II (Fig. 1). In contrast, H3K36me3 and H3K4me3 bound predominantly to upstream sequences and poorly to the 3′RR region. CTCF bound predominantly in the insulator region as previously described (7). Overall, the ERα and RNA Pol II binding patterns were best matched, illustrating a partnership of the two proteins within the immunoglobulin heavy chain locus.
As shown in Figure 2, when we added supplemental estrogen to purified, LPS-stimulated B cell cultures, ERα binding exhibited improved focus on the ERE hotspot in Sμ. RNA Pol II was similarly targeted to this site. Results supplement our previous discovery of synchronous shifts in ERα and RNA Pol II DNA binding patterns when supplemental estrogen was added to LPS-stimulated B cell cultures (47).
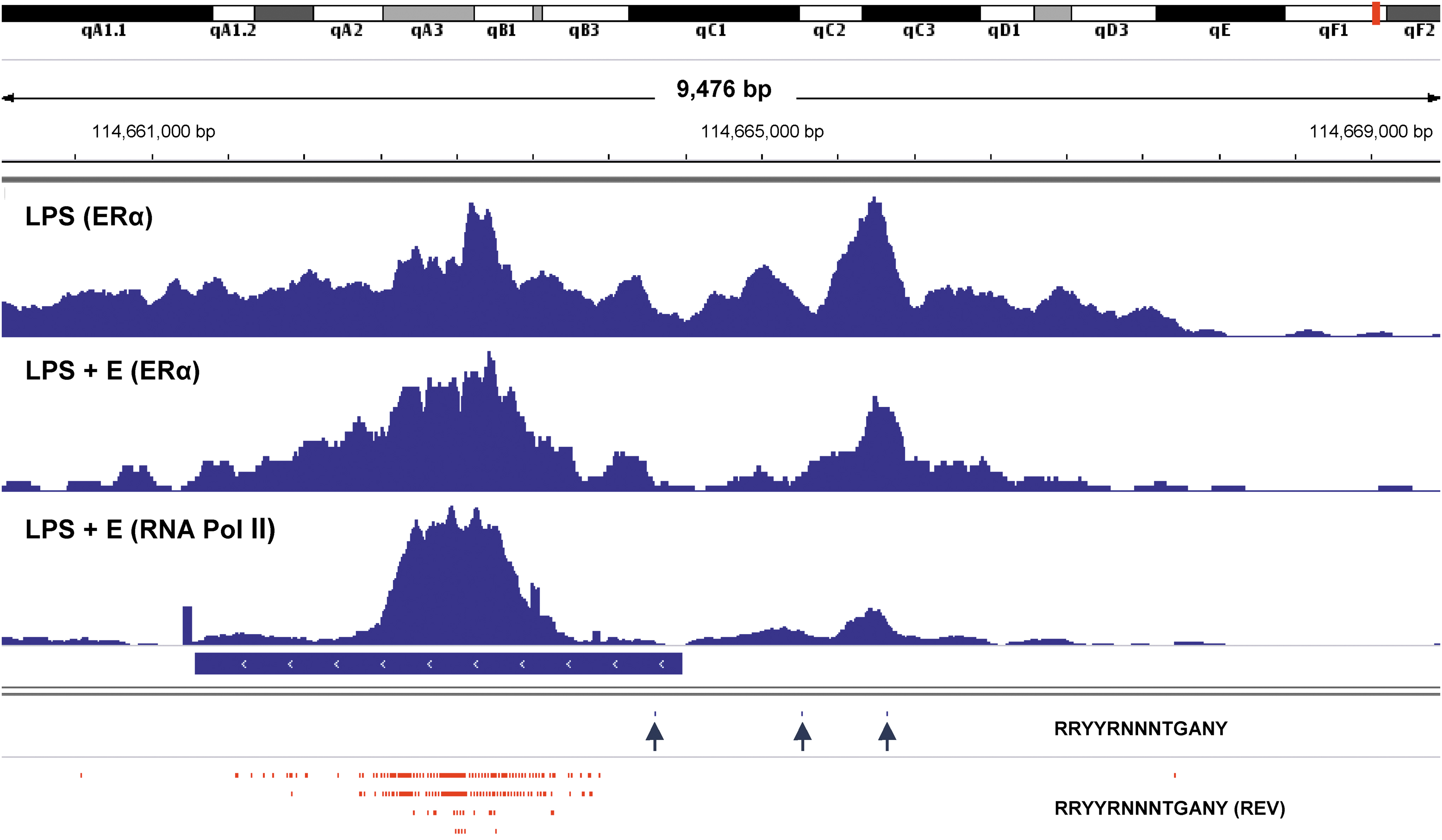
Focused binding of ERα and RNA Pol II on an ERE hotspot in Sμ in estrogen-supplemented B cell cultures. ChIP-seq libraries were prepared from purified B cells stimulated with LPS or LPS plus estrogen (LPS + E). In the latter case, both ERα and RNA Pol II were tested. Potential ERE (RRYYRNNNTGANY) were mapped using the IGV “Find Motif” function. ERE were identified in forward (blue ticks and arrows) and reverse (REV, red ticks) directions. The position of Sμ is indicated by a horizontal blue bar. Data ranges were 0–150 for the LPS library with ERα, 0–106 for the LPS + E library with ERα, and 0–233 for the LPS + E library with RNA Pol II. ERE, estrogen response element.
Focused binding of ERα to AC-repeats in the presence of supplemental estrogen
ERE hotspots (42) and individual ERE clearly mark some, but not all sites of ERα binding in the immunoglobulin heavy chain locus (46,47,79). We and others have previously identified AC-repeat sequences and other repetitive elements in and near the 3′RR among loci of rodents and primates (7,79,81,82). We therefore asked whether ERα binding associated with these sites (7,47) in B cells activated with LPS or LPS plus supplemental estrogen (LPS + E).
As shown in Figure 3, the ERα binding peaks were indeed aligned with AC-repeat sequences (mapped as “ACACAC” using the “Find Motif” function of IGV), particularly when B cells were stimulated in the presence of supplemental estrogen (LPS + E). AC-repeats are shown within the 3′RR in Figure 3 with blue and red ticks (indicating forward [left to right] and reverse (REV) sequence orientations, respectively). The four highest “LPS + E” peaks are indicated with arrows. Each peak aligned with at least one AC-repeat (at least 44 bases in length), either in a forward or reverse orientation. For example, the highest peak of ERα binding in Figure 3 (marked with a blue arrow) mapped to a sequence containing a 54 base AC-repeat straddled on both sides by one or two TGACC ERE half-sites; half sites were each located within 70 bases of the AC-repeat. We also observed focused ERα binding to AC-repeats and poly A sequences in and near Sμ, Cμ, and Cδ gene segments when supplemental estrogen was added to B cell cultures (45).
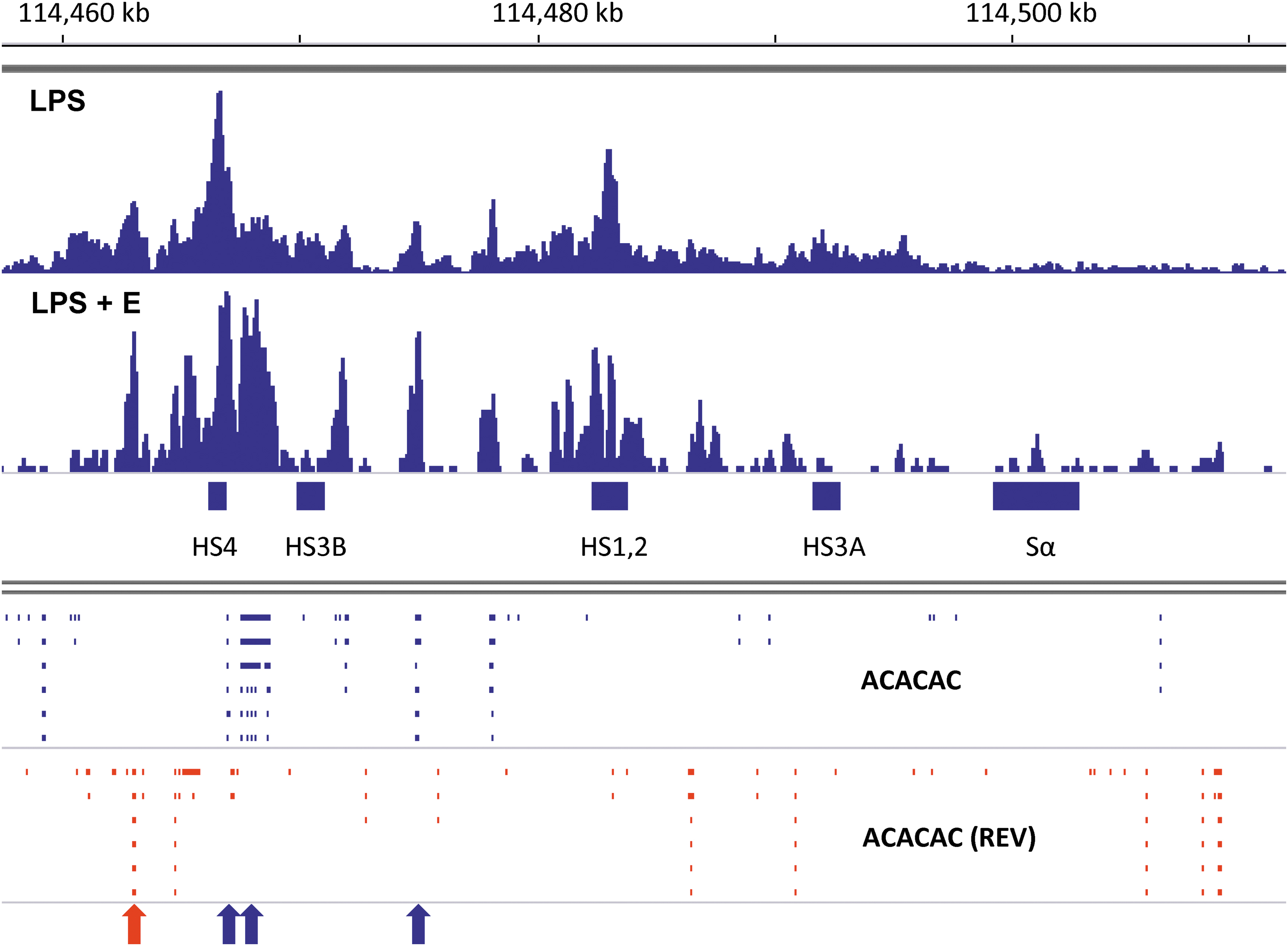
Shifts in ERα toward improved binding of AC-repeat sequences in the presence of supplemental estrogen. ChIP-seq libraries were from purified B cell cultures with LPS or LPS plus estrogen (LPS + E), shown using IGV software (mm9, chromosome 12). Data were normalized against 15M uniquely mapped reads (102). Data ranges were 0–134 for the LPS library and 0–87 for the LPS + E library. AC-repeat sequences (ACACAC) were mapped using the IGV “Find Motif” function. Ticks mark each sequence with a 5′–3′ orientation from left to right (blue) or right to left (red, termed “REV”). The four highest peaks of ERα binding in the LPS + E culture are marked with arrows, color-coded to match corresponding AC-repeat sequence orientations. AC, adenosine–cytidine.
Discussion
Synchronous binding of ERα and RNA Pol II
Data in this report reveal a striking similarity between patterns of ERα and RNA Pol II binding within the immunoglobulin heavy chain locus. Results supplement our previous finding that when estrogen was added to purified LPS-stimulated splenic B cells, there were synchronous shifts in binding patterns for ERα and RNA Pol II (47). Apparently, when estrogen ligands alter the conformation of ERα, both ERα and RNA pol II can be repositioned within the immunoglobulin heavy chain locus. These two transcription factors are clearly integral members of switchosomes (47,79) and enhanceosomes that regulate CSR and immunoglobulin expression patterns (99 –101).
In purified murine B cells, H3K27ac binding patterns were closely matched to those of ERα and RNA Pol II and H3K4me1 exhibited the next-best match. H3K36me3 and H3K4me3 associated predominantly with Sμ and surrounding regions, perhaps indicative of their roles in transcription initiation and elongation (33). The various histone marks will recruit different readers and thereby impact functions of RNA Pol II (2,33,35,59). CTCF, another factor known to regulate histone modifications and influence DNA architecture of the immunoglobulin heavy chain locus throughout B cell development, was associated with the HS5-8 insulator region as previously described (6,20,32). These proteins define only a fraction of enhanceosome composition, illustrating the enormous complexity of factors that influence enhanceosome function (7).
ER binds AC-repeat sequences, particularly when supplemental estrogen is added
We found that in the context of estrogen-supplementation, ERα binding to DNA was well focused on AC-repeats (Fig. 3). Sequence repeats were previously identified throughout the immunoglobulin heavy chain locus in both primates and rodents (79,82) and AC-repeats have been previously described as regulatory elements (41). These repeats are somewhat reminiscent of the heptamer-nonamer sequences instrumental in V-D-J joining during B cell development (e.g., a typical heptamer has the sequence CACAGTG). Possibly, for both V-D-J joining and CSR, ERα binding to AC-repeats assists DNA looping, alignment of DNA strands, and juxtaposition of regulatory elements, as is necessary for the initiation of DNA rearrangement events (26,30,98).
Cross-regulation of transcription factors
Transcription factors are cross-regulatory whereby changes in one hormone or transcription factor will alter the functions of others. As an example, as stated above, DNA loop formation is signaled by cooperative protein sets such as STAT5A, CEBPβ and PML or CTCF, RAD21, and SMC3 (9,95,103,104). Interactions have been described between ERα and NFκB (8,48,64), ERα and PPAR (49), ERα and STAT-5A (43,51,97), ERα and retinoic acid receptors (RAR) (55), and RAR and CTCF (43).
Nuclear hormone receptors compete both for DNA binding sites and ligands (39). This explains why patterns of antibody isotype expression are difficult to predict in vivo (3,24,39,48,51) and helps account for our previous finding that IgG2b is generally higher in C57BL/6 females compared to males, but that ratios can be reversed in the context of VAD (47). Perhaps estrogen supports IgG2b production, but ERα and RAR have competitive influences on CSR [vitamin A often drives the switch to IgA (55,77)]. If this is the case, estrogen's capacity to upregulate IgG2b in male mice may be more evident when vitamin A is absent.
From flu to lupus
As stated above, females and males respond differently to influenza virus (and other) infections and exhibit different frequencies of autoimmune disease. These differences are due, at least in part to variant estrogen levels, influenced by factors including sex, age, pregnancy, and hormone replacement therapies (34). We suggest that changes in estrogen and ERα binding to DNA may have profound influences on gene rearrangement and antibody output [as is the case for other factors such as Ikaros, Mediator, and the histone-reader bromodomain family member BRWD1 (62,80,92)]. Changes in antibody output may, in turn, translate to serious disease consequences (16,29).
Intentional and targeted manipulations of ERα binding within enhanceosomes and switchosomes of the immunoglobulin heavy chain locus (e.g., by using CRISPR-Cas9 technologies) (4,5,10,22–23,27,31,38,40,72,74,83,85,96), may eventually allow clinicians to improve control of pathogens and to reduce threats of autoimmune disease.
Conclusion
We previously identified hotspots for ERE in the immunoglobulin heavy chain locus, identified ERα binding to the locus, showed that estrogen induced synchronous shifts in DNA binding for ERα and RNA Pol II, and showed that deletion of ERE in HS1,2 or Eμ reduced CSR in a B cell line (7,46,47,79). Here, we show that ERα and RNA Pol II binding patterns within the immunoglobulin heavy chain locus have an extraordinary similarity and we show that ERα has a preference for binding to AC-repeat sequences in the 3′RR in the presence of supplemental estrogen. Data are presented to encourage further research to define functions of ERα and related nuclear hormones in the immunoglobulin heavy chain locus.
We emphasize that the binding of nuclear hormones to regulatory elements defines just one of many mechanisms by which nuclear hormones influence pathogens and pathogen control in mammals. Important ERα binding sites are also situated in T cell receptor loci and among V, D, and J gene fragments (47). Next steps will be to employ new molecular technologies to modify ERE and ERα-DNA binding patterns in vivo (1,4,5,10,11,23,27,31,38,40,72,74,83,85,96). Better understanding and control of ERα-DNA binding in the immunoglobulin heavy chain locus may ultimately allow clinicians to improve immune responses in cases of immunodeficiency and reduce immune responses in cases of allergic reactions or autoimmunity.
Footnotes
Acknowledgments
We thank Scott Olsen for assistance with library production. We thank the ENCODE consortium, the ENCODE production laboratories, John Stamatoyannopoulos (UW) and Bing Ren (UCSD) for generating and sharing ChIP-seq data sets (ENCODE ENCSR000CBC, ENCSR000CBD, ENCSR000CBE, ENCSR000CBY, ENCSR000CBZ, ENCSR000CDJ, ENCSR000CFT, ENCSR000CGA).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by NCI P30 CA21765, the intramural research program of the National Institutes of Health, National Institute of Aging, and ALSAC.