
Abstract
Hepatitis C virus (HCV) is a well-known pathogen to establish chronic infection leading to end-stage liver disease. The destruction of liver tissues takes its roots under chronic inflammation and proinflammatory signaling in liver microenvironment. The viral proteins interact with certain pattern recognition receptors, including Toll-like receptors, activating the innate immune system to clear the virus. HCV achieves immune evasion through other mechanisms and induce a continuous inflammatory microenvironment via Kupffer cells and Hepatic Stellate cells. This promotes disease progression. The current study aims to elucidate that the role of Toll-like receptor 4 (TLR4) induced innate immune response in chronic inflammation in patients chronically infected with HCV. For this purpose, changes in downstream signaling cascade of TLR4 during chronic HCV infection using peripheral blood mononuclear cells of chronic HCV patients were studied. We found significant increase in expression levels of proinflammatory and profibrotic genes induced by TLR4 Myeloid differentiation factor 88 (MyD88)-dependent pathway between treatment naive and healthy controls, while no significant difference between the expressions of genes involved in TLR4 signaling was found between treatment responders and healthy controls. Interestingly, both TLR4 MyD88-dependent and -independent pathways were found to be operational in nonresponders to interferon treatment. This further strengthens the involvement of innate immune signaling as a leading factor in HCV-mediated liver disease progression and the role of TLR4 MyD88-dependent and -independent pathway in ensuring the conditions for chronic inflammation.
Introduction
Hepatitis C virus (HCV), a notorious member of the family Flaviviridae, has been estimated to have infected about 3% population of the world with ∼4 million new infections arising each year (31). With the high rates of associated morbidity and mortality, it is the single most common indication for liver transplants (11) and costs billions of dollars annually exerting huge burden on public and private sector health care.
In Pakistan, an estimated of around 9.4 million people (about 5.4% of the population) are infected by HCV (3,20,25,24). The immune-based therapy for viral elimination is effective in about 50% cases of genotype 1, 4, 5, and 6 and about 80% cases of genotype 2 and 3 (43,46,48,54), but still viral relapse is very frequent (55,56) and the therapy itself has serious side effects that range from flu-like symptoms to thrombocytopenia and suicidal tendencies (7,12,27,33). An estimated 10–20% of the patients develop cirrhosis, while 1–5% patients may develop hepatocellular carcinoma at some point in their life (42,62).
First line defence against foreign invaders in our body is the innate system. Toll-like receptors (TLRs) is a group of evolutionarily conserved set of pattern recognition receptors found in a wide array of organisms (53) on innate immune cells. Up till now 11 TLRs have been recognized, functioning as initiators of inflammatory responses which triggers signaling pathways causing transcriptional activation and synthesis of proinflammatory cytokines and other proteins that promote host defense (8,29,30). These cytokines in turn activate inflammatory cascade, recruiting macrophages and neutrophils to the site of insult (13,64).
TLR4 or CD284 was the first human toll-like protein to be discovered (19,38), encoded by TLR4 gene, located on chromosome no 9. Different body tissues such as liver, peripheral blood leukocytes, and spleen express TLR4 receptors (35,37). Numerous pathogen-associated molecular patterns can act as ligand to activate TLR4 like gram-negative bacteria's lipopolysaccharides (LPS), taxol of plants, phosphorylcholine excreted by various Filarial nematode etc. (29,30). TLR4 can get stimulated by endogenous ligand like fibronectin, Tamm-Horsfall glycoprotein, and heparan sulfate of host as well (15,23,49). When ligand attaches to receptor, activation of TLRs leads to recruitment of immune cells like macrophages, NK cells, and other lymphocytes at the site of infection (9,49,65).
TLRs at their integrated side include a TIR domain which is Toll-IL-1 domain, conserved among all TLRs, which modulate TLR signaling. Proteins directly downwards of TLRs are TIR domain-containing adaptor-inducing IFN-β (TRIF), TRIF-related adaptor molecule (TRAM), Myeloid differentiation factor 88 (MyD88), MyD88 adaptor-like protein (MAL), all these also contain the same TIR domain motif, to interact with TLRs (44). TLR4 signaling occurs through two pathways: MyD88-dependent and TRIF-dependent pathways (1) (Fig. 1).
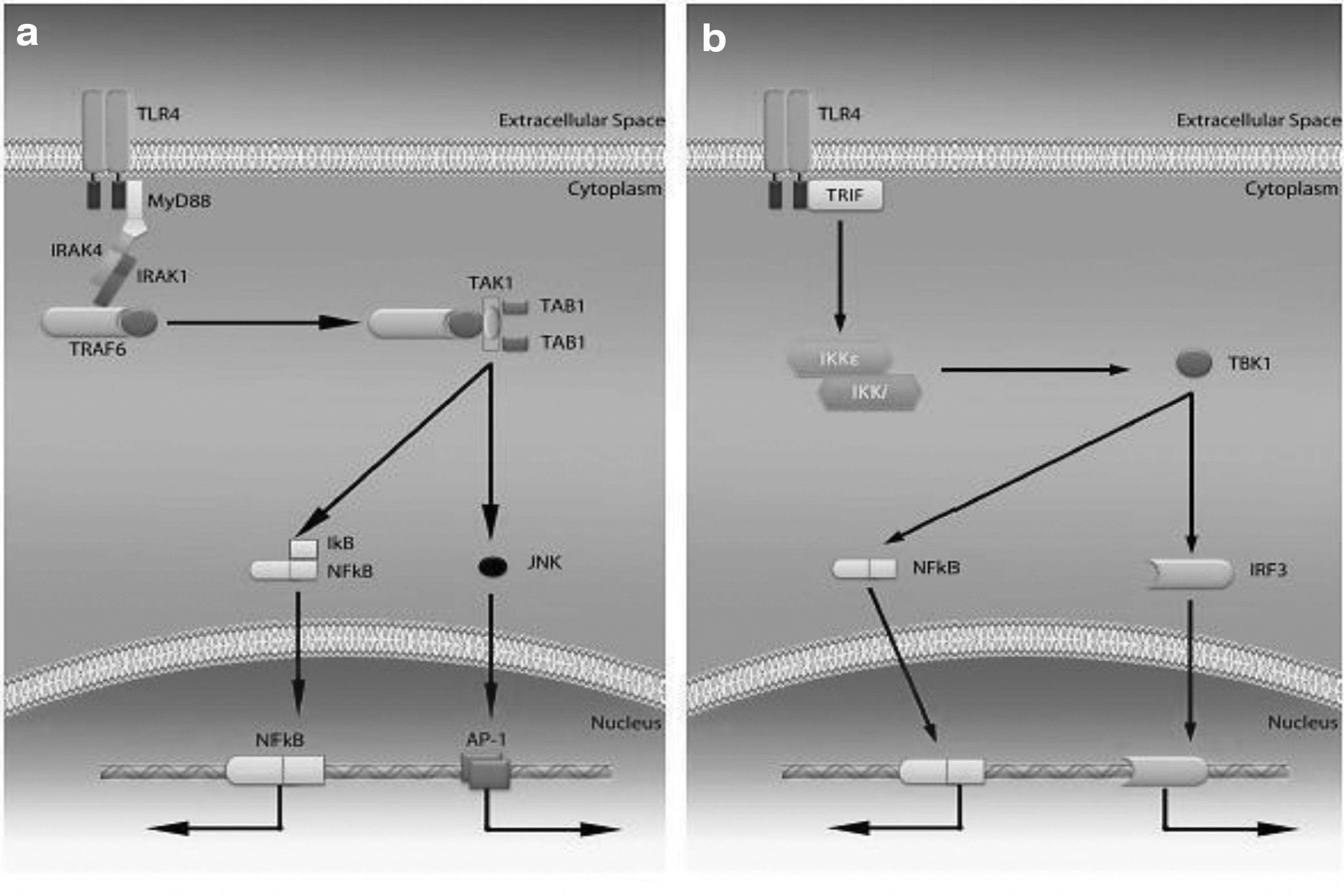
Injured liver cells having higher level of mRNA of TLR4, CD14, MD2, and other adaptor proteins causes the initiation of inflammatory gush mediated by TLR4 in the insulted organ (15,34,36). However, overexpression of TLR4 also shows deadly consequences in injured organ. Ex vivo studies on cell lines also suggested that during HCV infection, the TLR3 and TR4 get activated in liver sinusoidal endothelial and Kupffer cells, activate IFN-inducible genes, which then leads to strong and effective repression of the viral replication (6,32,64).
TLR4 acts as a double edge sword in HCV infection; during acute phase of HCV infection, it significantly upregulates pathways involved in viral elimination, but it turns into a bad guy during chronic HCV infection. In chronic patients of HCV, a high expression of TLR4 and other proinflammatory cytokines is reported along with the higher number of T regs, which varies with HCV subtype and its viral count in the host (39,64). This elevated expression of TLR4 and other cytokines along with the infiltration of immune cells, causes the liver cell damage, and in some cases leads the cells toward transformation into cancerous cells thus leading to hepatocellular carcinoma (42,61). TLR4 has been notoriously pronounced one out of seven genes that shows higher threat of cirrhosis development in chronic HCV-infected patients (17,24,30).
TLR4 and its downstream targets can essentially be used as a potent therapeutic target to dampen HCV-associated liver cell damage in chronic HCV patients, especially in known interferon (IFN) therapy nonresponders. In developing countries, such as Pakistan, direct acting antivirals (DAAs) are being used in clinical practices to treat treatment naive patients, relapse cases due to viral reactivation of postsustained virological response (SVR), and previously known nonresponders to IFN and ribavirin standard therapy (26). Some of the poor treatment naive patients are also given the option of less expensive alternate to DAA; the standard IFN and ribavirin therapy, in case their medical is not covered by the government-based fund. Therefore, ribavirin and IFN therapy is still important and relevant in developing countries (20,21,47,66).
This study was designed to evaluate TLR4 signaling in innate immune response against HCV infection in patients who have undergone IFN and ribavirin therapy compared with treatment-naive and healthy controls and its role in induction of profibrotic microenvironment via proinflammatory signaling.
Materials and Methods
Clinical sample collection and ethical approval
Peripheral blood samples from HCV-infected patients with written informed consent were collected in 3 mL EDTA vacutainers from Armed Forces Institute of Pathology (AFIP), Rawalpindi, Pakistan, and transported to Atta-ur-Rahman School of Applied Biosciences (ASAB), National University of Sciences and Technology (NUST) Islamabad. Ethics Committee approval was obtained from both institutes. The ethical approval includes IRB approval for clinical sampling from AFIP and IRB to deal with samples from NUST.
The selection criteria for inclusion and exclusion were basis of patient's medical history. Patients with coinfection of hepatitis B virus, Epstein Bar Virus, cytomegalovirus and tuberculosis were excluded from the study. HCV-positive patients with any history of renal injury, hypertension or cardiac malfunctioning, hypo/hyperthyroidism, rheumatoid arthritis, asthma, and diabetes were also excluded from study to avoid any bias caused by inflammation response in metabolic or autoimmune disorders. Patients who were under DAA treatment were also excluded from the study, since the aim of the study was limited to study the TLR4 signaling in patients that were treated with IFN therapy in the past and were either nonresponders or relapse cases post-SVR for at least 6 months. This limited the included sample size to 30 HCV-infected cases only.
HCV-infected patients included in the study were divided into 3 subcategories, with 10 individuals in each subgroup category. The subcategories were devised on the status of IFN-α and ribavirin treatment. Treatment-naive patients were newly diagnosed with chronic HCV infection and had not received any antiviral treatment. Those who had previously received a standard IFN-α and ribavirin combinational treatment were regarded as responders and nonresponders based on active detectable viral infection after 24 weeks of treatment course.
Ten healthy blood donors were selected randomly from Armed Forces Institute of Transfusion (AFIT), Rawalpindi, who have undergone thorough screening for infections, as healthy controls for comparison. All samples were immediately processed within 2 h of collection. Baseline clinical information is summarized in Figure 2 and Table 1.
Whisker's plot, portraying age limit of study groups, with age at y-axis and categories at x-axis; central line showing the median while marginal lines showing the range limit.
Mean Age ± Standard Error of Mean and Median Age of Patients in Three Study Groups and Control Group
Isolation of peripheral blood mononuclear cells from clinical samples
Peripheral blood mononuclear cells of 10 control subjects and 30 HCV patients were isolated by density gradient centrifugation, using Ficoll-Histopaque (Histopaque®-1077; Sigma-Aldrich), according to manufacturer's guideline. The pellets obtained were gently washed with phosphate-buffered saline and preceded for RNA extraction.
RNA extraction and complementary DNA synthesis
RNA extraction of total RNA via TRIzol® Reagent (catalog number: 15596-026; Invitrogen, CA) was performed, according to manufacturer's instructions. Extracted RNA was treated with DNase I (catalog number: EN0521; Fermentas, ON, Canada) to circumvent any DNA contamination. Furthermore, RNA was quantified using Eppendorf BioPhotometer Plus (Eppendorf, NY) and was run on 1% agarose gel to confirm the integrity total extracted RNA.
The complementary DNA (cDNA) was then synthesized using RevertAid H minus Reverse Transcriptase (Fermentas) and Oligo-dT 18 primer, plus RiboLock™ RNase Inhibitor (Fermentas). Confirmation of synthesized cDNA was done using polymerase chain reaction (PCR) of housekeeping gene that is, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene.
Quantification of targeted genes
Complete coding sequences of targeted genes from NCBI gene bank were used to design primers by using Primer 3 software. Similarity index of each primer was analyzed individually both in BLAST and BLAT to avoid common occurring sequences of human genome and requested from the operon, commercially manufacturing distributors at HVD Biotech Vertriebs GmbH Wurzbachgasse 18 (Wurzbachgasse, Vienna, Austria) (Table 2 and Fig. 3).
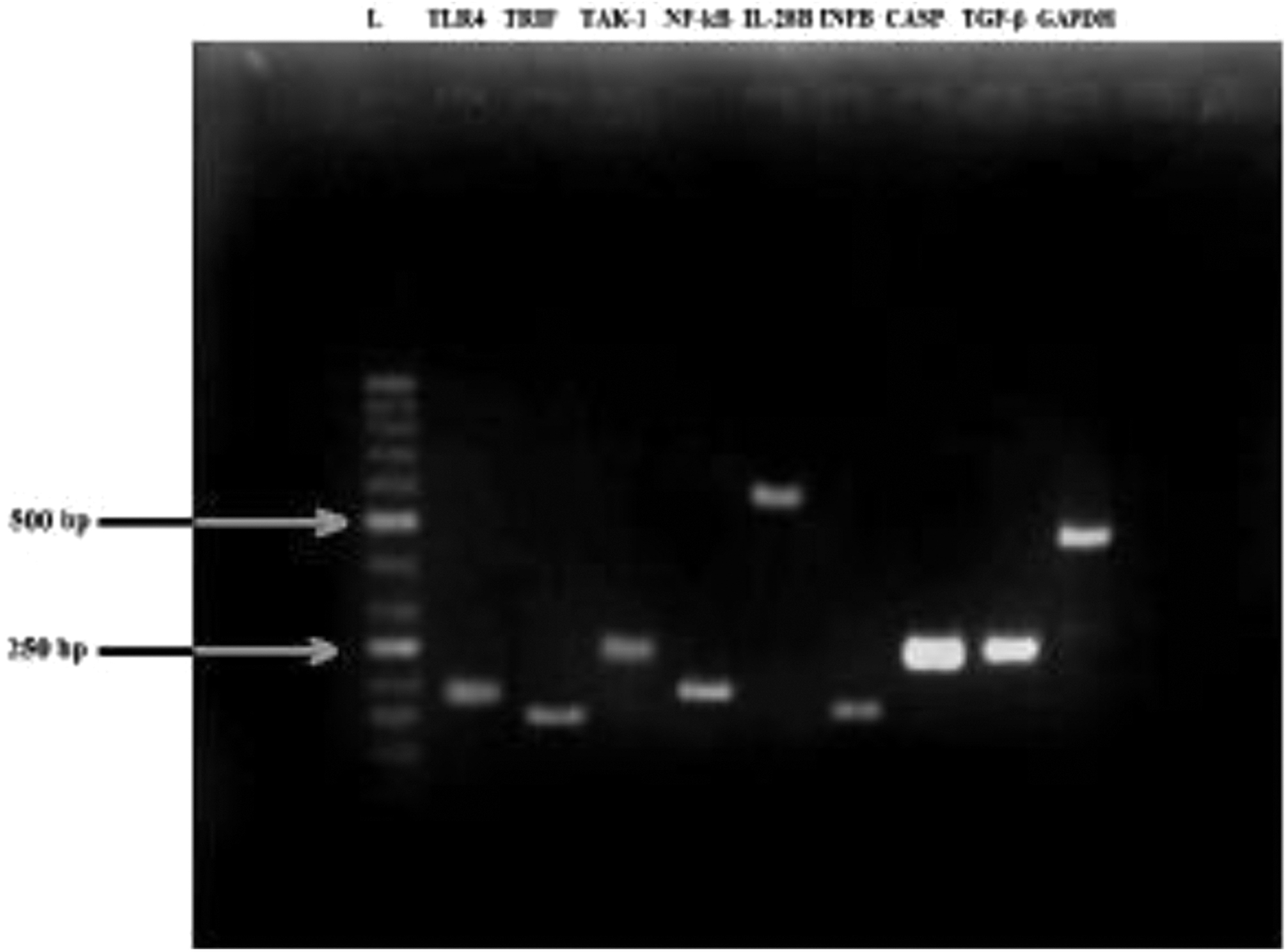
Gel electrophotogram of 2% Agarose gel showing polymerase chain reaction amplification products of TLR4 (198-bp), TRIF (165-bp), TAK-1 (248-bp), NF-κB (198-bp), IL-28B (552-bp), IFN-B (152-bp), CASP 1 (245-bp), TGF-β (240-bp), and GAPDH (453-bp) (house-keeping gene) at annealing temperature of 60°C, (L = ladder 50 bp). GAPDH, glyceraldehyde 3-phosphate dehydrogenase
Primer Sequences and Their Product Sizes Used to Amplify Genes of Interests Both for Conventional and Quantitative Real-Time Polymerase Chain Reaction
The mRNA levels of TLR4, TRIF, TAK1, NF-κB, IL-28B, IFN-β, Caspase1, and TGF-β were determined by quantitative real-time PCR (qRT-PCR). Using Maxima® SYBR Green/ROX qPCR Master Mix (2 × ) (catalog number: K0221; Fermentas), targeted genes were amplified on Applied Biosystems 7300 Real-Time PCR System. The reactions were preheated to 95°C for 10 min, then were run for 40 cycles at 95°C (denaturation) for 15 s and 60°C (annealing) for 60 s. Quantified expression of selected genes in chronic HCV samples of all categories in comparison with healthy controls was done using cDNA. Fold change in expression profile of genes was calculated by first normalizing, cycle threshold (Ct) values of selected genes by comparing with GAPDH (reference housekeeping gene). All experiments were performed in duplicates.
Statistical analysis
Statistical analysis was performed using GraphPad prism, version 6.0 (GraphPad Software, CA). Test applied was Student's unpaired t-test where p ≤ 0.05, p ≤ 0.005, and p ≤ 0.001 were considered statistically significant.
Results
Transcriptional analysis TLR4 and downstream targeted adaptor proteins in treatment naive compared to healthy controls
A significant increase of 1.3-fold (p < 0.05) was observed in the expression of TLR4 gene along with a significant increase of 2-fold (p < 0.01) in TAK-1 expression, while no significant change was observed in TRIF levels in treatment naive compared to healthy controls. Expression of NF-κB and its downstream anti-inflammatory cytokines during chronic HCV infection in treatment naive patient group was also compared to healthy controls. A significant increase of 1.8-fold (p < 0.05) was observed in the expression of NF-κB gene along with a significant increase of 1.4-fold (p < 0.05) in expression of IL-28B and a 1.8-fold (p < 0.01) increase in expression of TGF-β was observed in treatment-naive group, when compared to healthy controls. No statistically significant difference of expression of INF-β and caspase 1 was observed between the two groups (Fig. 4a).
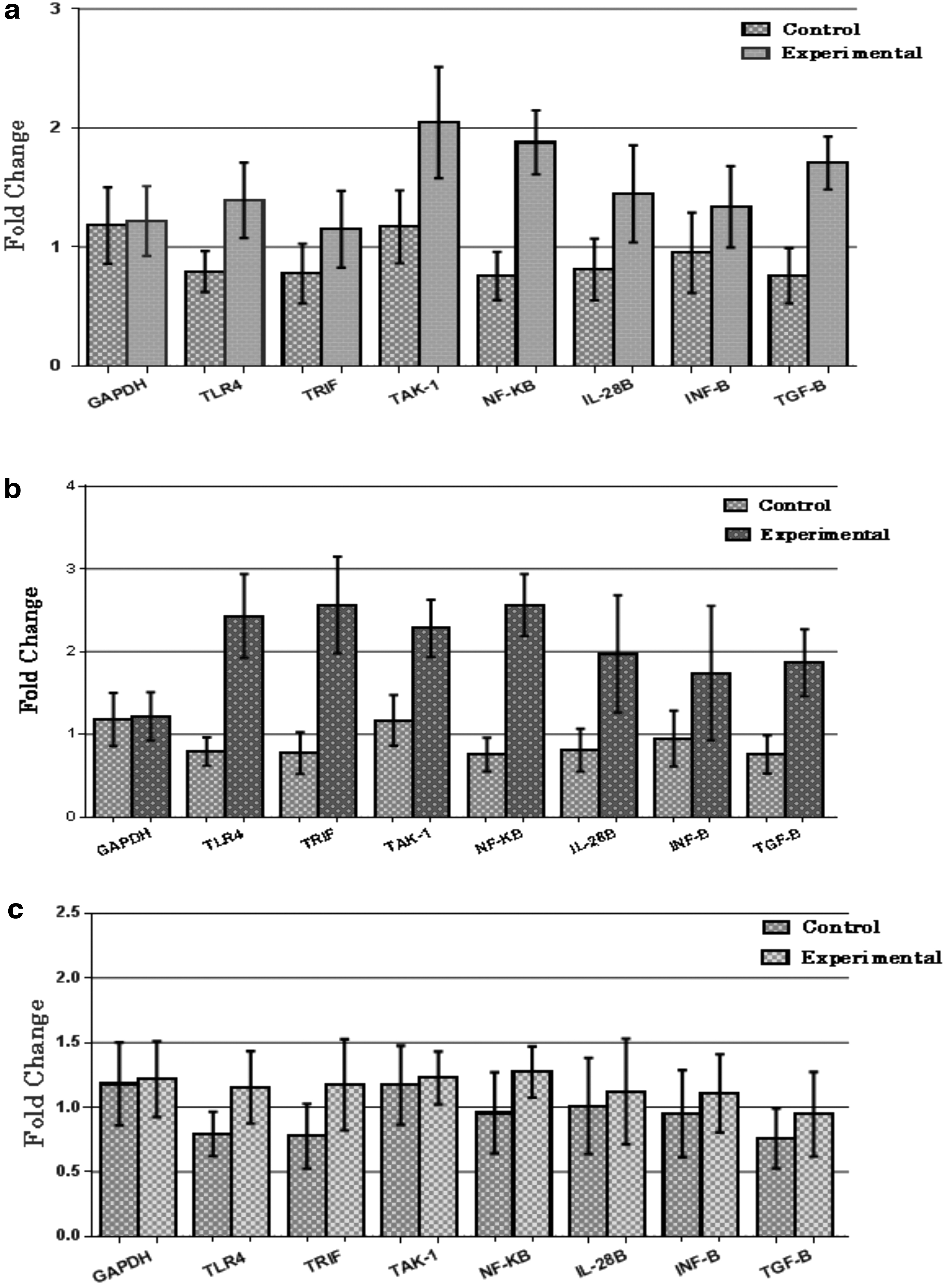
Real-time quantitative expression in terms of fold change of desired genes, in peripheral blood mononuclear cells of chronic HCV patients (experimental) when compared with healthy individuals (controls).
Transcriptional analysis TLR4 and downstream targeted adaptor proteins in treatment nonresponders compared to healthy controls
A significant increase of 2.3-fold (p < 0.001) was observed in the expression of TLR4 gene along with a significant increase of 2.2-fold (p < 0.01) in TAK-1 expression, while TRIF expression was increased by 2.5-folds (p < 0.01) in treatment nonresponders, when compared to healthy controls. A significant increase of 2.4-fold (p < 0.001) was observed in the expression of NF-κB gene along with a significant increase of 2-fold (p < 0.01) in expression of IL-28B. Moreover a significant increase of 1.7-fold (p < 0.05) in IFN-β and 1.8-fold (p < 0.05) of statistically significant increase in TGF-β expression was observed (Fig. 4b). Caspase 1 remained unchanged between treatment nonresponders and healthy controls.
Transcriptional analysis TLR4 and downstream targeted adaptor proteins in treatment responders compared to healthy controls
No significant change was found in any of the gene expressions of TLR4, TRIF, TAK 1, NF-κB, INF-β, TFG-β, IL-28B, and caspase 1, between the treatment responders and healthy controls (Fig. 4c). All values were expressed as ±SEM, p < 0.05 for experimental versus control by using unpaired Student's t-test.
Discussion
A significant role is played by Toll-like receptors in mounting innate and adaptive immune response against the attack of any foreign pathogen. Allele variations among the TLR genes have also been implicated in disease severity, disease prognosis, and treatment prognosis (14,18,37,41,56). According to many studies, the HCV proteins interact directly with the host proteins to perturb the signaling pathways (8,10,11,19,28,40).
Liver is an important organ of the body and can also be considered as a chief entity for the bacterial products residing in the gut system. An increase in pace of bacterial translocation has been reported in many cases, where different models of liver disorders were investigated. LPS is one of the expected candidates for TLR4-dependent fibrosis initiation. Moreover, many current researches have also reported that activation of TLR4 can be carried out by certain endogenous ligands, including HMGB1 and Hyaluronan as well (23).
One of the significant traits of HCV infection is impaired cellular signaling. This impaired signaling is known to cause steatosis, cirrhosis, and even hepatocellular carcinoma (HCC) in some extreme cases (4,10,17,52,59,63). Moreover, some of the proteins of HCV, for example, NS3 protease and the core protein, are known to hinder the downstream signaling proteins that are, Stat1, IRF-3, TRIF, TBK1, and Cardif, ultimately restraining IFN signaling (5,8,11,28,35,52,61).
According to many studies, an acceleration of liver disease in HCC occurs in presence of higher expression of TLR4. In addition, polymorphism in TLR4 gene and other TLRs has also been linked with the severity of the disease and treatment outcome (14,18,37,57). The interaction between viral protein NS5A with promoter region of TLR4 gene has also been established (52). Therefore, it is further suggested that LPS from the intestine and the NS5A protein, both interact with the promoter regions of TLR4, resulting in its upregulation of TLR4 signaling treatment-naive patients. Based on established role of TLR4, the study was designed to investigate the differential expression of TLR4 and adaptor proteins involved in chronic HCV infection.
Our results confirmed that MyD88-dependent pathway, and not TRIF, is upregulated in treatment-naive patients, when compared with healthy controls. MyD88-dependent TLR4 signaling pathway operates through Tak-1 complex, consisting of TAK1 and the adapter proteins TAB1 and TAB2 (60). TAK-1 expression was found to be elevated in both treatment-naive patients and treatment nonresponders, when compared to healthy controls, confirming the role of MyD88-dependent TLR4 signaling in chronically infected patients. This activation of TAK-1 by TLR4 can further lead to the activation of transcriptional factor NF-κB. In turn, NF-κB signaling alters the liver microenvironment to proinflammatory and profibrotic condition via upregulation of many inflammatory cytokines (both pro and anti) such as IL-6, IFN-α, IL-10, IL-28b, IL-29, etc., and TGF-β.
Viral proteins such as NS5A and NS3 are involved in upregulation of NF-κB via LPS-dependent mode of action and by augmenting ROS and ER stress, eventuating the peril for insulin resistance, carcinoma, and inflammation (8,13,28,36,45,52). We too observed statistically significant increase in NF-κB signaling, resulting in upregulation of downstream IL-28B as anti-inflammatory marker and TGF-β as profibrotic marker in treatment naive and treatment nonresponders, when compared with healthy controls.
MyD88-independent TLR4 signaling was observed in treatment nonresponder patients as the expression of TRIF was found to be elevated in comparison to healthy controls. However, no prominent change in mRNA expression level of TRIF was observed in treatment-naive patients. We can conclude that the INF-α in combination with ribavirin therapy somehow triggers the second downstream signaling pathway of TLR4 in treatment nonresponders. However, no significant difference in TLR4 signaling was observed in treatment responders, when compared with healthy controls, posttherapy. It is reported in literature and is a well-known fact that the activation of TRIF sparks two separate lineages of molecular events. The first one is late onset of NF-κB by RIP1 and TRAF6; while, the second lineage occurs via TRAF3, which then leads to the dimerization and thus activation of IRF3 (INF regulatory factor 3). IRF3 then moves inside the nucleus and act as transcription factor for INF-β (16,22). This was confirmed by the upregulation of INF-β in treatment nonresponders, in comparison to healthy controls.
The upregulation of TGF-β was observed in both treatment naive and treatment nonresponders with respect to healthy controls. The role of TGF-β induced collagen deposition in extracellular matrix is well established in the typical fibrotic liver (4,17,50). Although the primary source for TGF-β is considered to be Kupffer cells in paracrine signaling (52), it is also reported to be produced in autocrine manner by hepatic stellate cells. In hepatic stellate cells, upregulation of collagen I, II, and IV is established by TGF-β signaling. This upregulation leads to transdifferentiation of myofibroblasts in naive stellate cells (2,51). TLR4-dependent inflection of TGF-β signaling grants a link between proinflammatory and profibrogenic signals. It is previously reported that, in TLR4 knockout mice, the level of fibrogenesis was notably reduced. Moreover, mutant mice strains which have mutation in their CD14 or LPS gene also showed reduced hepatic fibrosis compared to their active counter parts (23).
Based on our study and known mechanism, we speculated that the upregulation of TLR4 signaling (MyD88 dependent and independent) might lead to downregulation of pseudoreceptor BAMBI of TGF-β via autocrine and paracrine signaling between hepatic stellate cells and Kupffer cells (19,58,64). Therefore, TLR4 activation and upregulation of NF-κB is responsible for liver tissue fibrosis during chronic infection of HCV in treatment naive and treatment nonresponders, but not in treatment responders.
Caspase 1expression analysis was included in the study, based on its stimulation by NLRP3 inflammasome in parasitic infections via TLR signaling. MyD88-dependent TLR signaling pathway upregulates NF-κB expression, which as a transcription factor, results in production of pro-IL-1B precursors and other inflammasome inducible structures. However, we found no significant difference in expression of caspase 1 in between the experimental groups and control group. Therefore, we were unable to confirm this mechanism of acute inflammation via TLR4 signaling in chronic HCV infection (8).
Conclusion
In current study, the activation of TLR4 pathway in chronically HCV-infected patients under combinational therapy (INF-α and ribavirin) and treatment naive was compared with healthy control. Upregulation of TLR4 expression was observed in treatment naive and treatment nonresponders, but not in treatment responders and healthy controls. Furthermore, an enhanced expression of profibrotic marker, such as TGF-β, along with upregulation of IFN-β and IL-28B genes in treatment nonresponders in comparison with healthy controls. It can be stated that during HCV infection, TLR4 plays a dual role, by upregulating expression of several anti-inflammatory genes like IL-28b to mount an antiviral response on one hand, but on the other hand, it also drives disease progression by inducing liver cell damage over the extended periods of time.
Further investigations into the role of highly expressed TLRs signaling cascades during HCV infections shall help us understand inflammation and disease progression. Moreover, relevant studies should be projected to evaluate the role of TLR4 signaling in disease risk stratification and prognosis in treatment-naive patients. Our study provides the foundation and proposes the use of TLR4 inhibitors in patients, who were previously resistant to INF-α and ribavirin combinational therapy and treatment-naive patients, along with DAAs, to reduce liver tissue damage.
Footnotes
Authors' Contributions
S.M. conceived the idea and designed the study. S.K. executed the experiments and did the analysis under guidance of S.M., M.A.M., and K.S. wrote the article. S.M. critically reviewed and revised the article. S.M. being principal investigator facilitated S.K. in smooth conductance of all experiments.
Ethical Approval
Ethical Approval for sampling was attained by Armed Forces Institute of Pathology (AFIP), Rawalpindi, Pakistan, and Armed Forces Institute of Transfusion (AFIT), Rawalpindi. The ethical approval for molecular work on clinical in BSL 2 facility was obtained from ASAB, NUST, Islamabad, Pakistan.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding received from any source.