
Abstract
In retroviral infections, different immunological mechanisms are involved in the development of a chronic infection. In the Friend virus (FV) model, regulatory T cells (Tregs) were found to induce CD8+ T cell dysfunction before viral clearance is achieved and thus contribute to viral chronicity. Although studied for decades, the exact suppressive mechanisms of Tregs in the FV model remain elusive and an unavailable therapeutic target. However, extracellular IL-2 and intracellular NF-κB signaling were shown to be important pathways for Treg expansion and activation. Therefore, we decided to focus on these two pathways to test therapeutic approaches inhibiting Treg activation during FV infection. In this study, we show that the inhibition of either IL-2 or the NF-κB subunit c-Rel, impaired Treg expansion and activation at 2 weeks post-FV infection. Total numbers of Tregs as well as activated Tregs were reduced in FV-infected mice after treatment with anti-IL-2 antibodies or the c-Rel blocking reagent pentoxifylline. Surprisingly, this did not affect the expansion or function of virus-specific CD8+ T cells nor viral loads in the spleen. However, our data suggest that neutralization of IL-2 as well as blocking c-Rel efficiently inhibits virus-induced Treg expansion.
Introduction
When the mammalian immune system is challenged with a retrovirus, T cells play a major role in virus control. While CD4+ T helper cells induce inflammation and T cell recruitment, CD8+ effector T cells mediate killing of infected host cells (45). The CD4+ T cell population also includes regulatory T cells (Tregs), which act anti-inflammatory during an immune response and maintain self-tolerance (40).
By using the Friend virus (FV) mouse model [reviewed in Dittmer et al. (13)], it was shown for the first time that Tregs play a pivotal role in virus immunity (12,13,21). On one hand Tregs counter-regulate effector T cell responses preventing immunopathology during viral infections, but at the same time they interfere with virus clearance contributing to chronic infection (19). During the late phase of acute FV infection Tregs expand and induce CD8+ T cell dysfunction, thereby preventing viral clearance (50,51,53). We could show, that a short-term ablation of Tregs during chronic FV infection improved the antiviral activity of CD8+ T cells with a subsequent decrease in viral loads (9,10). There is evidence that Tregs also play an important role in T cell dysfunction during HIV and simian immunodeficiency virus (SIV) infection, but whether this is beneficial or detrimental in lentiviral infections is a matter of current investigations (19,27,28,31,32,36).
Although investigated for years, the exact immunosuppressive mechanisms of Tregs during FV infection remain elusive, hence inaccessible as therapeutic targets (19). Therefore, possibilities to control the expansion and activation of Tregs have become interesting therapeutic approaches. Up to now, we could only interfere with FV-induced Treg expansion in transgenic DEpletion of REGulatory T cell (DEREG) mice, in which Tregs express the diphtheria toxin receptor and can be experimentally depleted by injection of the corresponding toxin (9). Although these experiments revealed very important information about the function of Tregs in vivo (19), they did not provide clues on how to therapeutically target the expansion of Tregs in humans.
Tregs are known to be highly dependent on IL-2 signaling for their activation and proliferation (4,14,30), which has also been shown for the FV model (33). Interestingly, during FV infection there is a minor subpopulation of TCR Vβ5+ Tregs that expands independently of IL-2 signaling, whereas the vast majority of Tregs is IL-2-dependent (24,33). Thus, we suggest that the neutralization of IL-2 prevents Vβ5− Treg expansion and might attenuate subsequent CD8+ T cell exhaustion.
In addition to the direct effect of IL-2, it was shown that NF-κB signaling is important for IL-2 expression in CD4+ T helper cells and Foxp3 expression in Tregs (8,15,20,29,39,46,47). The NF-κB subunit c-Rel was identified as key protein in this pathway and its inhibition led to an impaired suppressive capacity of Tregs in a mouse tumor model (16,35). This was discovered with c-Rel-deficient mice and verified by therapeutic applications of pentoxifylline (PTXF), an FDA-approved drug targeting and blocking c-Rel (16,34,48). Thus, we wanted to test whether PTXF could be used to inhibit Treg suppression during a retroviral infection in the FV mouse model.
Based on our previous findings with Treg ablation in DEREG mice and its impact on CD8+ T cell functionality as well as the ability to reduce viral loads, we investigated possibilities to impair Treg expansion during an ongoing infection by using anti-IL-2 (αIL-2) antibodies or PTXF in a therapeutic manner.
Materials and Methods
Mice and infection
Female C57BL/6 mice (Envigo, Rossdorf, DE) with an age of at least 8 weeks at experimental onset were used. Mice were infected with the FV complex containing B-tropic Friend murine leukemia helper virus and polycythemia-inducing spleen focus-forming virus. The stock was prepared as 15% spleen cell homogenate from susceptible BALB/c mice infected for 14 days with 3,000 spleen focus-forming units of FV. Mice were infected i.v. with 20,000 spleen focus-forming units of FV. The stock was free of lactate dehydrogenase virus. Mice were sacrificed at 14 days postinfection (dpi) by cervical dislocation.
Animal experiments were performed under strict consent with the German regulations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). North Rhine-Westphalia State Agency for Nature, Environment and Consumer Protection (LANUV) approved all experiments and protocols. Some of the experiments have been performed in the Department of Infectious Diseases, Union Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, P.R. China. These animal experiments were performed under strict consent with the Chinese and Hubei province regulations.
In vivo IL-2/c-Rel inhibition
For IL-2 inhibition mice were i.p. injected with an αIL-2 mAb mixture consisting of 50 μg anti-IL2 clone JES6-1A12 and 50 μg anti-IL2 clone S4B6 (BioXCell) in 500 μL phosphate buffered saline (PBS). c-Rel was depleted by the i.p. injection of 1.25 mg PTXF (Sigma-Aldrich, St. Louis, MO) in 500 μL PBS.
Flow cytometry
Single cell suspensions were prepared from spleens. CD8+ T cell and Treg markers were defined as previously described (9,23). Cells were stained with the following antibodies: CD4 (GK1.5), CD8 (53-6.7), CD43 (1B11), CD45.1 (A20), CD107a (1D4B), Foxp3 (FJK-16s), granzyme B (GzmB, GB11), Helios (22F6), IFNγ (XMG1.2), IL-2 (JES6-5H4), Ki-67 (SolA15), KLRG1 (MAFA), TNFα (MP6-XT22), TNFR2 (TR75-89), Vβ5 (MR9-4). The antibodies were purchased from BioLegend (San Diego, CA), eBiosciences (San Diego, CA) or BD Biosciences (San Jose, CA). FV-specific CD8+ T cells were stained with PE-coupled MHC class-I H-2Db tetramers containing the F-MuLV GagL 85-93 peptide (Abu-Abu-L-Abu-L-T-V-F-L) (MBL, Woburn, MA). Dead cells were excluded from the analysis by using a fixable viability dye (eFluor780; eBiosciences). For restimulation, 96-well flat bottom MaxiSorp plates were coated with anti-CD3 (145-2C11) in sodium carbonate at 4°C overnight. Subsequently, the cells were incubated with anti-CD28 (37.51) and Brefeldin A at 37°C, 5% CO2 for 5 h. Cells were fixed with BD Cytofix/Cytoperm (BD Biosciences) or Foxp3/Transcription Factor Fixation/Permeabilization kit (eBiosciences) according to the manufacturer's instructions. Data were acquired at LSR II flow cytometer (BD Biosciences). Data analysis was done using FlowJo 10.5 software (Tree Star, Inc., Ashland, OR).
Infectious center assay
To determine infectious centers, single cell suspensions of splenocytes from infected mice, were cocultured with Mus dunni cells at 37°C and 5% CO2 for 3 days. The samples were fixed with 96% ethanol, washed with PBS and bovine serum albumin (PBS + BSA) and stained with F-MuLV envelope-specific mAb 720 (37). Finally, the cells were incubated with a peroxidase-conjugated goat anti-mouse antibody (Dako, Hamburg, Germany) followed by an aminoethyl carbazole substrate (Sigma-Aldrich) to visualize the foci that originated from infected cells.
In vivo cytotoxicity assay
The in vivo cytotoxicity assay was performed using donor cells of CD45.1 congenic C57BL/6 derived splenocytes, loaded with the F-MuLV CD8+ T cell peptide Abu-Abu-L-Abu-L-T-V-F-L (peptides&elephants, Henningsdorf, Germany). Peptide-loaded and CFSE-stained (BioLegend) cells as well as unloaded and unstained cells were mixed in a 1:1 ratio and 3 × 106 cells of this mixture were injected i.v. in 14 days FV-infected mice 1 h before sacrificing them. The cells were stained and analyzed as stated earlier. The cytotoxic capacity was determined by the normalized quotient of CFSE+ and CFSE− CD45.1+ cells, detected in the spleen of the corresponding recipient mouse.
Statistical analyses
Statistical analyses were done with GraphPad Prism 6 (GraphPad Software, San Diego, CA). Statistical differences between groups of FV-infected and either naive or treated mice were examined by Mann–Whitney U test (nonparametric).
Results
To investigate the possibilities of therapeutic Treg inhibition during FV infection, we infected wild-type C57BL/6 mice with FV and started either αIL-2 or PTXF daily therapy 8 dpi (Fig. 1A). The animals were sacrificed at 14 dpi and the T cell populations were analyzed by flow cytometry.
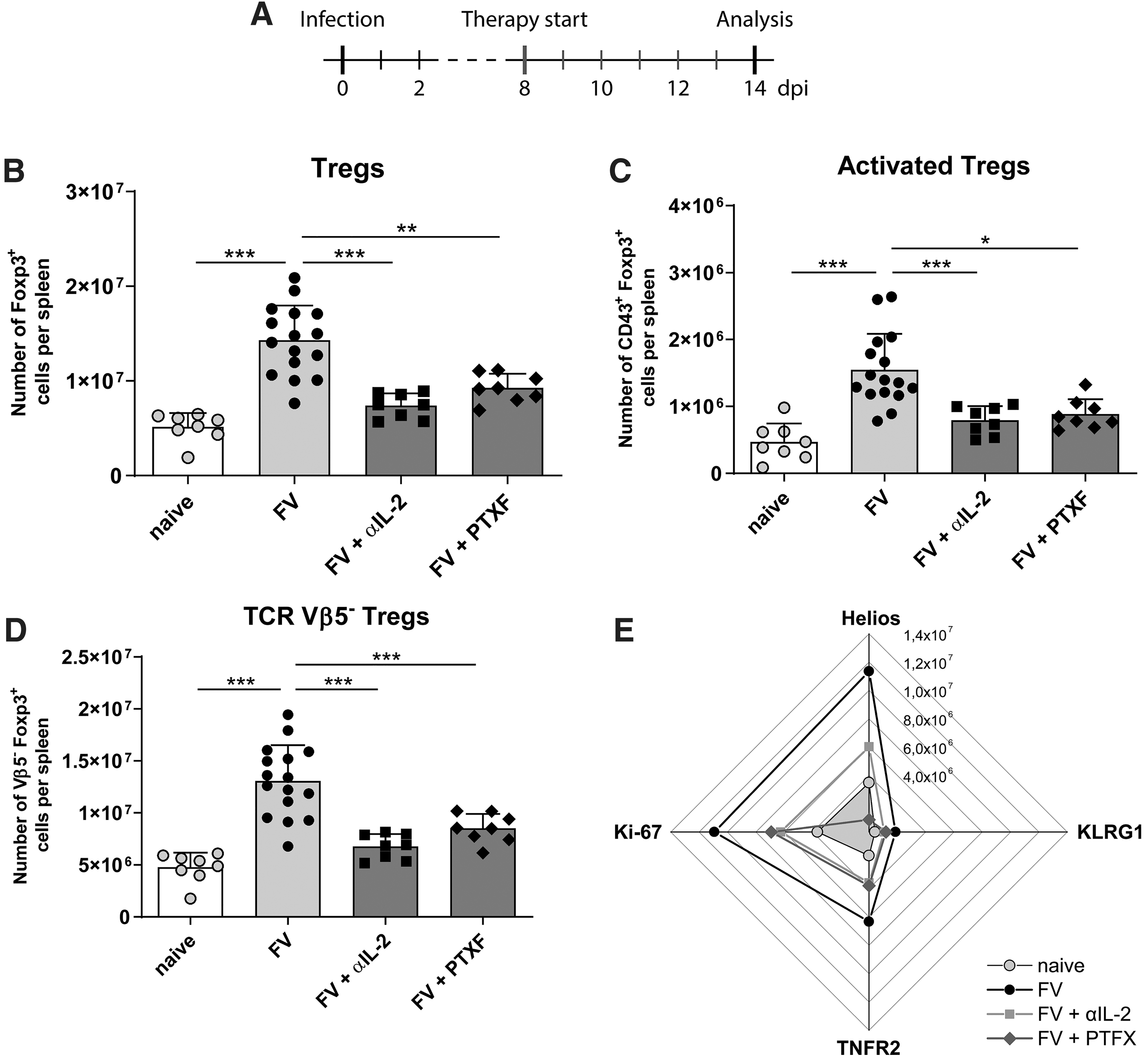
Treg expansion and activation in the spleen of FV-infected mice after αIL-2 or PTXF therapy. Mice were infected with FV, received a daily therapy with αIL-2 or PTXF (8–14 dpi) and were finally analyzed by flow cytometry (14 dpi)
Expansion and activation of Tregs are reduced after αIL-2 and PTXF therapy
In the first step we analyzed the total numbers of Tregs per spleen in naive FV-infected but therapy-naive and FV-infected treated mice. As previously shown (51), the numbers of Tregs in the spleen of FV-infected mice were significantly higher compared with naive mice, demonstrating the substantial expansion of Tregs upon FV infection. In both groups of treated mice this Treg expansion was no longer observed. Total numbers of Tregs did not differ between naive and αIL-2-treated mice; therefore, the Treg numbers were significantly lower compared with FV-infected therapy-naive mice. Similar findings were obtained for the numbers of Tregs in PTXF-treated mice. Treg numbers were significantly higher compared with naive mice, but at the same time they were significantly lower compared with therapy-naive FV-infected mice (Fig. 1B). However, after both therapies the Treg levels were still higher compared with naive mice. When analyzing total numbers of activated (CD43+) Tregs per spleen we found significantly more activated Tregs in the spleen of infected but therapy-naive mice compared with αIL-2- or PTXF-treated mice. These numbers were in the same range as in naive animals (Fig. 1C). Thus, both therapies, targeting Treg activation and proliferation, were able to efficiently interfere with Treg expansion in vivo during a retroviral infection.
An important aspect was the analysis of the numbers of TCR Vβ5− Tregs, since they are known to be IL-2 dependent in FV infection. We found that the total numbers of Vβ5− Tregs were increased upon FV infection in the spleen of mice, an effect that was abrogated in the treated groups (Fig. 1D). Thus, both therapies prevented the expansion of IL-2-dependent Tregs.
Finally, we analyzed the influence of the therapies on the Treg activation marker Helios, the maturation marker KLRG1, the functional marker TNFR2 and the proliferation marker Ki-67. The numbers of Tregs expressing these markers were all reduced by αIL-2 or PTXF therapy compared with therapy-naive infected mice and were similar to naive mice. Only Helios was an exception, because the numbers of Tregs expressing this activation marker, also suggested to be a marker for thymus-derived Tregs (1,17), were clearly reduced in αIL-2-treated mice, but still about two to four times higher compared with PTXF-treated or naive mice (Fig. 1E).
Summarized, the total numbers of IL-2-dependent Tregs as well as activated or proliferating Tregs were significantly reduced when both therapies were applied, indicating the potential of IL-2 neutralization or NF-κB block in inhibiting Treg responses. However, these effects were not observed in the bone marrow (data not shown), where both therapies had surprisingly no impact on Treg numbers. Therefore, we focused on the spleen for the following analyses of CD8+ T cells.
CD8+ T cell expansion and activation remain unaffected after αIL-2 and PTXF therapy
The next step was to investigate the effect of the therapy-impaired Treg response on the expansion and activation of CD8+ T cells. We measured total numbers of CD8+ T cells and virus-specific effector T cells as well as functional properties of CD8+ T cells after therapy.
Compared with naive mice, the total numbers of CD8+ T cells were significantly higher in FV-infected therapy-naive mice, but did not differ significantly in either αIL-2- or PTXF-treated animals (Fig. 2A). Surprisingly, also the numbers of activated (CD43+) effector and virus-specific CD8+ T cells were not affected by both therapies and were comparable with FV-infected therapy-naive mice (Fig. 2B, C).
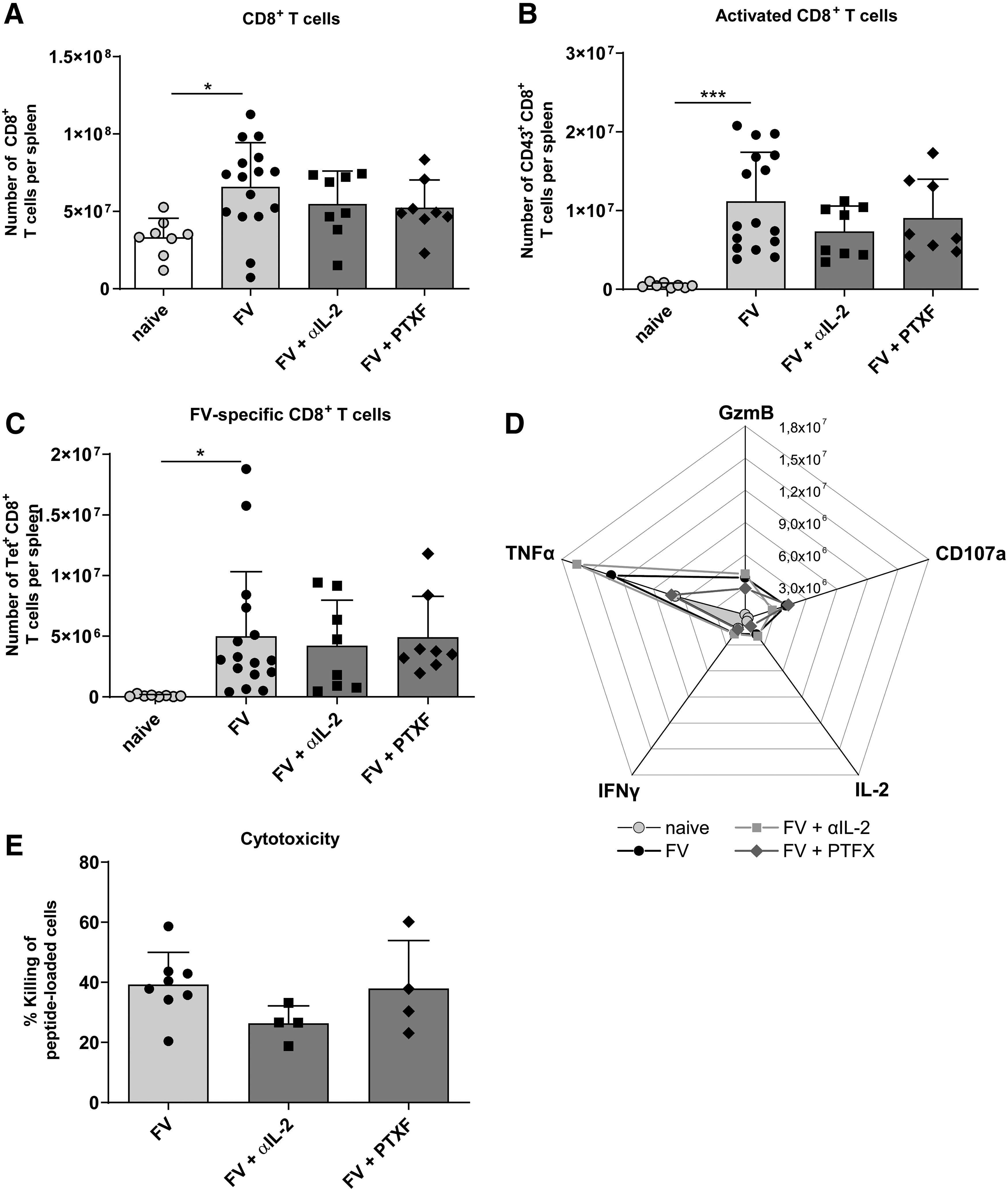
CD8+ T cell activation and expansion in the spleen of FV-infected mice after αIL-2 or PTXF therapy. Mice were infected with FV, received a daily therapy with αIL-2 or PTXF (8–14 dpi) and were finally analyzed by flow cytometry (14 dpi). The total numbers of all CD8+ T cells
Furthermore, also functional markers that are typical for an effector phenotype of CD8+ T cells were unaffected by both therapies. This was true for numbers of CD8+ T cells expressing the cytotoxic molecule GzmB, the degranulation marker CD107a as well as the cytokines TNFα, IFNγ, and IL-2 (Fig. 2D).
By using an in vivo cytotoxicity assay, we quantified the killing of FV peptide loaded target cells through CD8+ T cells. As shown in Figure 2E, the cytotoxic capacity was similar in treated and untreated FV-infected mice. Taken together, Treg responses were substantially suppressed by αIL-2 or PTXF therapy, whereas the number and functionality of virus-specific CD8+ T cells remained unchanged.
Virus control is not influenced by αIL-2 and PTXF therapy
The final step was to analyze the impact of the therapy on the viral loads and spleen weights in treated mice. As shown in Figure 3A, the viral loads did not significantly differ between the therapy-naive and therapy-positive groups. Also spleen weights, which can be increased by FV infection (5), were not affected by any therapy (Fig. 3B). These results matched with the shown CD8+ T cell functions that define the control of acute FV infection fundamentally (11,18,25,38). Therefore, the desired therapeutic effect was not achieved.
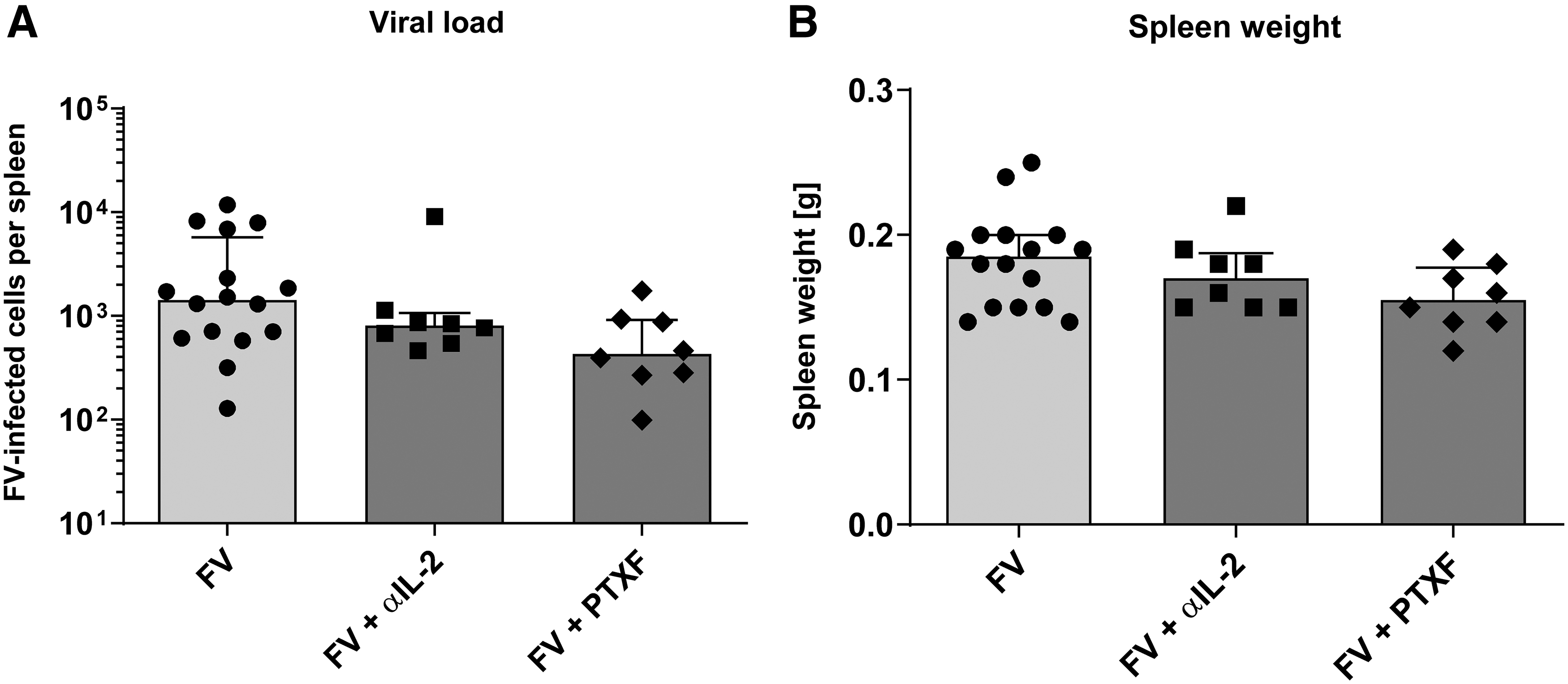
Viral loads and spleen weights of FV-infected mice after αIL-2 or PTXF therapy. Mice were infected with FV, received a daily therapy with αIL-2 or PTXF (8–14 dpi), and were analyzed by an infectious center assay (14 dpi). The viral loads
Discussion
In this study, we demonstrated that the expansion and activation of Tregs upon FV infection can be inhibited, when the infected mice received either an αIL-2 or PTXF therapy. The overall numbers of Tregs, as well as the numbers of activated, matured, and proliferated Tregs were significantly reduced when the mice received these therapies. This was in line with findings of Myers et al. (33) and Grinberg-Bleyer et al. (16). Of note, this effect did not fully reproduce the effect of Treg depletion in DEREG mice (9), as a residual amount of Tregs was left after αIL-2 or PTXF therapy.
Therapy did not affect the CD8+ T cell response in either case, which remained similar to that of untreated FV-infected mice. This was a surprising result, as Tregs are considered to be an important suppressive factor of CD8+ T cell responses in many infections, and their depletion in DEREG mice was shown to improve CD8+ T cell functions in the FV model (9).
These studies revealed an increased number of CD8+ T cells expressing IFNγ, TNFα, and IL-2 after Tregs were completely ablated (9). These cytokines are produced by CD8+ T cells during acute FV infection and contribute to the viral control (22,33,44,50). During the late phase of acute FV infection, the production of cytokines is decreasing and CD8+ T cells become dysfunctional (50,52). In our experiments, the therapies did not alter the number of CD8+ T cells expressing any of these cytokines, except for TNFα in PTXF-treated mice. This was in line with previous findings showing that TNFα expression is suppressed by PTXF (41).
Besides cytokines, CD8+ T cells produce cytotoxic molecules, for example, granzyme A and B as well as perforin, to kill FV-infected cells (51,52), but also lose this ability when becoming dysfunctional (50,52). Again, in the current experiments, the numbers of CD8+ T cells expressing cytotoxic molecules were unchanged after Treg-targeted therapy and in vivo killing of target cells, loaded with an FV-specific peptide on MHC I (51), were not affected. Overall, although the number of Tregs was reduced significantly, this did not affect these hallmarks of CD8+ T cell functionality, contrary to former experiments in DEREG mice (9).
In fact, reduced Treg responses that did not result in boosting effector functions of CD8+ T cells are shown here for the first time in FV research. Thus, it has to be discussed how these unexpected findings could be explained. Possibilities are that both treatments did not only affect Treg responses, but also negatively interfered with robust CD8+ T cell activity or that residual Tregs were still sufficient to suppress CD8+ T cells.
IL-2 is known as an important signal molecule for CD8+ T cells (3). Although priming of effector CD8+ T cells is mostly IL-2 independent (49) the proliferation, maturation, and survival of CD8+ T cells require IL-2 (2,6). CD8+ T cells upregulate the high-affinity IL-2 receptor CD25 after priming and respond to IL-2 that is mainly produced by CD4+ helper T cells or activated CD8+ T cells (50). Not surprisingly, IL-2 consumption is one of the suppressive mechanisms of Tregs targeting CD8+ T cell proliferation and function (43). Thus, IL-2 neutralization successfully reduced the numbers of IL-2 dependent Tregs, but at the same time, it also may have limited the proliferation and effector cell maturation of virus-specific CD8+ T cells. Such opposing therapy effects maybe resulted in a very similar magnitude of the CD8+ T cell response compared with nontreated control mice and no improved control of virus replication. In contrast to the DEREG model, the current therapy did not deplete Tregs completely. In fact, Treg numbers were higher in FV-infected treated animals than in naive mice. Therefore, it must be taken into consideration that residual Tregs may still be able to sufficiently suppress the CD8+ T cell activity. Hence, the ultimate goal of our therapeutic approach, an improved control over chronic retroviral infection, could not be achieved by IL-2 neutralization.
Unexpectedly, the PTXF therapy had also no effect on CD8+ T cell responses, although the numbers of activated and matured Tregs were significantly reduced. Our findings with the PTXF therapy appear contrary to the results of Grinberg-Bleyer et al. (16), who reported that the inhibited suppression by Tregs led to improved tumor control. Importantly, they had to combine PTXF therapy with anti-PD-1 antibodies to achieve a significant decrease in tumor growth, whereas PTXF alone was ineffective. Recently, we also showed the importance of PD-1 signaling in CD8+ T cell functionality in the FV model (7). However, in this study, we did not test a combination therapy with PTXF and anti-PD-1 antibodies.
Furthermore, c-Rel was shown to inhibit the IL-2 expression in T cells (26,42). This important cytokine might have been missing for a more effective CD8+ T cell response, similar to what we discussed for IL-2 neutralization. It is possible that in both therapeutic approaches the reduced numbers of Tregs were dependent on IL-2 inhibition. In addition, the design of our therapy experiment might also be involved in the therapy outcome. In contrast to our treatment starting at 8 dpi, Grinberg-Bleyer et al. (16) started the PTXF therapy 1 day before tumor challenge. As a consequence, our experimental settings might have failed to completely interfere with the Treg activity.
Another interesting observation was that the profound effect of both therapies on Tregs was only observed in the spleen, but not in the bone marrow. This was surprising, since we previously showed that Tregs infiltrate, mature, and proliferate in the FV-infected bone marrow (51), which likely also requires IL-2. If our experimental drugs did not reach the bone marrow or if Tregs are more IL-2 independent at this site of infection remains unknown. However, the results indicate that activated Tregs were still present in protected sites of animals receiving αIL-2 or PTXF therapy, with the possibility that they subsequently influenced CD8+ T cell responses in the spleen.
In summary, we were able to inhibit the expansion of Tregs during the acute phase of FV infection by therapy with αIL-2 neutralizing antibodies or by inhibiting the NF-κB subunit c-Rel with PTXF. Nevertheless, this did not change the functionality of CD8+ T cells nor the control of viral replication. However, the profound effect on Tregs is a promising finding that should be further investigated to develop new tools for the manipulation of Treg responses in chronic viral infection.
Footnotes
Authors' Contributions
J.A.R. and U.D. designed, and J.A.R. performed the experiments, analyzed the data, and performed statistical analyses. A.M. was involved in sample preparation. A.M., L.O., and J.L. commented on the article. U.D. and J.L. conceived the study. J.A.R. and U.D. wrote the article. All authors read and approved the final article.
Data Availability Statement
All data sets generated and/or analyzed during this study are available from the corresponding author on reasonable request.
Acknowledgments
We thank Elisabeth Littwitz-Salomon for the critical discussion of the experiments.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Deutsche Forschungsgemeinschaft (DFG) (DFG DACH DI714 17-1 and DFG DI714 20-1).