
Abstract
Acute viral infections are characterized by rapid increases in viral load, leading to cellular damage and the resulting induction of complex innate and adaptive antiviral immune responses that cause local and systemic inflammation. Successful antiviral immunity requires the activation of many immune cells, including T cells, natural killer cells, and macrophages. B cells play a unique part through their production of antibodies that can both neutralize and clear viral particles before virus entry into a cell. Protective antibodies are produced even before the first exposure of a pathogen, through the regulated secretion of so-called natural antibodies that are generated even in the complete absence of prior microbial exposure. An early wave of rapidly secreted antibodies from extrafollicular (EF) responses draws on the preexisting naive or memory repertoire of B cells to induce a strong protective response that in kinetics tightly follows the clearance of acute infections, such as with influenza virus. Finally, the generation of germinal centers (GCs) provides long-term protection through production of long-lived plasma cells and memory B cells, which shape and broaden the B cell repertoire for more effective responses following repeat exposures. In this study, we review B cell responses to acute viral infections, primarily influenza virus, from the earliest nonspecific B-1 cell to early, antigen-specific EF responses and finally to GC responses. Throughout, we address known factors that lead to distinct B cell response outcomes and discuss how their functions effect viral clearance, highlighting the critical contributions of each response type to the induction of highly protective antiviral humoral immunity.
Introduction
Acute viral infections are responsible for some of the most terrifying epidemics on record, with the 1918 influenza virus pandemic killing at least 50 million worldwide (54). Today, the threat of influenza virus-mediated pandemics is still looming, with billions of domesticated and wild animals acting as reservoirs for multiple strains of influenza capable of undergoing point mutations, as well as reassortment between strains, changes that allow for both seasonal influenza outbreaks in humans and the potential for more deadly pandemics.
Ebola virus (EBOV), one of many emerging viruses causing acute infections, has caused many recent outbreaks in Western and Central Africa and is associated with gruesome clinical symptoms and high mortality rates (1). While these outbreaks have remained relatively isolated due to immense efforts in establishing quarantines, and through education in exposure prevention, population growth and increased urbanization rates enhance the interface between humans and the natural reservoirs of these viruses. This in turn further increases the risk of outbreaks and make containment ever more challenging.
While endemic viruses such as influenza and emerging viruses such as EBOV differ greatly in genetics and pathophysiology, similarities can be drawn in terms of viral replication kinetics and timing of clinical symptom onset. Complete protection against these viruses depend on multiple factors: The timely generation of antiviral antibodies that are specific for viral components, which either directly neutralize the virus by inhibiting the invasion of new host cells or promote antibody-dependent virus clearance through Fc receptors, as well as natural killer (NK) cells and CD8 T cells that clear virus by killing infected cells. It was shown that CD8 T cells are dispensable for influenza virus clearance when strong and rapid T-dependent antibody responses were induced (32). The presence of antibodies is particularly critical also for protection upon secondary challenge, as circulating and local respiratory tract antibody production can prevent reinfections. These antibodies appear to offer also at least partial protection against strains of influenza virus that are distinct (heterosubtypic) from those that caused a previous infection (85). Strong heterosubtypic immunity generated against pandemic strains of influenza provide a broad level of population or “herd” immunity, dampening morbidity and mortality rates of phylogenetically related pandemic strains. This was seen during the last pandemic in 2009, during which the presence of antibodies to previous pandemic H1N1 strains provided partial protection against a new triple reassortant H1N1 “swine-flu” virus (44,136).
During primary EBOV infection, early, antiviral antibody responses have been associated with protection (10) and, along with a strong inflammatory response, are highly correlated with asymptomatic infections, clearance, and survival (69,70). The data indicate that antibodies are also critical in containing rapidly replicating EBOV and that they can prevent virus-induced death. Therefore, understanding how B cells are activated during acute viral infections to generate highly effective and long-lasting antibody responses, and the mechanisms by which they contribute to virus neutralization and clearance, could guide the development of more effective vaccines that induce broadly protective and long-lasting antiviral immunity.
In this study, we review the distinct B cell responses induced by viral infections and the contribution of B cell subsets to these responses. We primarily focus on influenza infection in mice, but reference other viral infection models and data when appropriate. We also describe recent research into the activation and differentiation of distinct B cell subsets with model antigens where necessary, to integrate the known mechanisms of antibody response development with those obtained from viral infections.
Naturally Occurring Antibodies Generated by B-1 Cells Provide Early Protection from Viral Infection
Spontaneously produced and circulating polyreactive “natural” antibodies (natAbs), mostly consisting of secreted immunoglobulin M (sIgM), are required for optimal immunity to acute viral infections (13,88). Serum natural IgM levels were shown to correlate with survival from influenza virus infection and early virus replication (13). Of note, these systemic levels of natural IgM did not change following acute influenza infection (24). This is consistent with the fact that most natAbs are produced independent of exposure to foreign antigens by populations of fetal- and neonatal-derived and activated B lymphocytes, hereto referred to as B-1 cells (112,140). Unlike the majority of bone marrow-derived “conventional” follicular B cells (B-2 cells), which are eliminated when binding to self-antigens during their development, positive selection for relatively strong self-reactivity appears to be a required step in the development of B-1 cells (46,47). While this explains the self-reactive nature of natAbs, how the population of self-reactive B-1 cells is prevented from causing antibody-mediated autoimmune disease is an open question, as is the question of how this selection process seems to select for natAbs that can bind and neutralize numerous pathogens. It is the reactivity to these pathogens, however, that enables natAbs to form a strong and nonredundant first line of preexisting adaptive immune defense, as summarized in greater detail elsewhere (123).
While natural IgM can bind to and neutralize a variety of influenza A and B strains (12), the exact viral epitopes have not been elucidated. It is interesting to consider that, as an enveloped virus, influenza virus uses host cell membrane in viral protein and genome packaging thus containing both host- and virus-derived antigens. It is well established that antiphospholipid antibodies are part of the natAb repertoire and that they recognize host cells undergoing changes in cell membrane composition during events such as apoptosis (37). Thus, it is conceivable that early detection and neutralization of enveloped viruses may rely on the natAb self-specificity for dead/dying cells rather than the detection of viral antigens per se. Of importance, the presence of IgM has been shown to enhance activation of complement induced by apoptotic cells, which in turn would likely lead to enhanced phagocytosis of dead and dying cells, which can reduce viral propagation (45). Together it suggests that natAb, particularly IgM, can provide enhanced protection through both viral neutralization, as well as through enhanced removal of viral infected cells.
Regulation and Function of natAb Production by B-1 Cells
Several heterogenous subsets of spontaneous natAb-secreting B-1 cells have been identified in spleen and bone marrow, but not the peritoneal and pleural cavities; the latter are sites where many nonsecreting B-1 cells reside (25). natAb-secreting B-1 cells in bone marrow and spleen are B-1 derived plasma cells (B-1 PCs) and “classical” B-1 cells, respectively (112). natAb production, similar to production of antigen-induced antibody, was reported to be dependent on the master regulator of antibody secretion, Blimp-1 (115). Indeed B-1 PCs have a very similar phenotype (IgM+, CD19lo_neg, CD138+) and gene expression signature (Blimp-1hi, IRF-4hi) as fully-differentiated B-2-derived plasma cells, and lack of Blimp-1 expression by B cells reduces natAb production by that cell subset (112). This is consistent with significant reductions in serum IgM levels (113). The remaining IgM and most steady-state immunoglobulin G (IgG)3 production, however, seemed to be generated by nonterminally-differentiated B-1 cells (112). This is similar to a report about Blimp-1 dependent and -independent IgM production in the shark (22). However, the mechanisms inducing and sustaining natAb production by these nonplasma cells remain to be identified. In response to influenza virus infection, B-1 PCs and B-1 plasmablasts also rapidly appear in the respiratory tract draining mediastinal lymph nodes (medLNs), where they secrete IgM for a few days, suggesting their activation and differentiation from antigen-stimulated B-1 cells (112). However, only roughly 10% of the B-1-derived IgM produced in the medLN can bind to influenza virus (24). What stimulated the other cells to secrete IgM is currently unknown, but, as outlined below, was recently shown to require expression of toll-like receptors (TLRs) by B-1 cells (111).
Existing evidence suggests that continuous “tonic” B cell receptor (BCR) signaling is an important regulator of natAb production, as it regulates the extent to which B-1 cells are stimulated to differentiate into B-1 PC in spleen and bone marrow. Changes to these signals alter natAb levels and therefore antiviral immunity (87). One example is expression of the BCR signaling-inhibitor CD5 by the majority of B-1 cells, as well as by self-reactive anergic B-2 and self-reactive T cells (15,117). Expression of CD5 inhibits activation of B-1 cells following BCR-crosslinking (121), indicating that CD5 regulates natAb production by inhibiting overshooting activation of B-1 cells through BCR-mediated self-antigen recognition. In addition, changes in signaling through the costimulatory molecule CD19 have also been shown to suppress BCR-mediated activation of B-1 cells (28). Furthermore, chronic self-antigen stimulation of B-1 cells was shown to result also in increased Nur77 expression, which in turn constrained natAb production (51). Consistent with those findings, Nur77 knockout mice (Nr4a −/−) had significantly increased levels of natAbs compared to controls (50). Programmed death ligand-2 (PD-L2) has also been shown to play a role in controlling the amount of autoreactive natAbs, as expression of PD-L2 by B-1 cells reduced natAb levels (78).
Finally, expression of the Fc-receptor for sIgM (FcμR) (93) was shown to regulate the extent of IgM-BCR expression on B cells, by regulating the transport of the IgM-BCR to the B cell surface during development (86). Global or B cell restricted deletion of the FcμR−/− enhanced the levels of natural IgM, which enhanced control of early influenza replication in the lungs of infected mice (86). Together these data strongly indicate that BCR signaling controls the levels of natAb production and, by so doing, controls early viral replication. It is likely that the beneficial effects of enhanced B-1 cell activation must be balanced with the increased risk of autoimmunity; as in most of the above instances, enhanced natAb production was correlated with increases in autoantibody production.
natAb provides immune protection likely by a number of distinct mechanisms. In addition to direct neutralization of the virus, natAbs can enhance the binding of complement to influenza virus particles, which increases virion aggregation, effectively reducing the number of infective particles (52). Influenza virus neutralization mediated by natural IgM was shown to depend on activation of early complement factors (C1, C2, C3, C4), but independent of end-stage components of the complement pathway, suggesting indirect receptor-mediated means of viral clearance (52). Complement-receptor mediated binding can also enhance antigen presentation and therefore the induction of adaptive immune responses (34).
Indeed, natural IgM was shown to enhance influenza virus-specific IgG, and passive immunization with B-1 derived IgM rescued the virus-specific IgG responses in mice lacking sIgM (13). This contribution of natural IgM to the antigen-specific antibody response might be facilitated by many different mechanisms, including more efficient delivery of antigen to professional antigen-presenting cells (APCs) and follicular B cells, more efficient delivery of pathogen-associated molecular patterns to pattern-recognition receptors such as TLRs on APCs resulting in enhanced inflammation, and/or increased BCR signaling on B-2 cells through the recruitment of virus antigen by sIgM-FcμR binding. Importantly, the rapid production of IgM by conventional B cells should provide similar direct and indirect antiviral immune defense mechanisms as natural IgM, suggesting additional perhaps unique functions for B-1 cell-derived natural IgM that are yet to be revealed.
B-1 Cells Respond to Innate Signals to Produce the Earliest Antiviral B Cell Response
Given the above discussion on the inhibitory role of CD5 for B-1 cell responses, it was surprising that during infection with influenza virus, as well as other infections, the CD5+ and not the CD5− B-1 cells responded to the infectious insult, migrating from the body cavities to the regional lymphoid tissue, where they differentiated into antibody secreting B-1s and B-1 PCs (24,111,131,132). Analysis of the B-1 cell responses revealed that innate Type I interferon (IFN) receptor-mediated signaling was regulating the B-1 cell response. While local secretion of influenza infection-induced Type I IFN caused the CD11b-dependent accumulation of B-1 cells in the regional lymph nodes (LNs) (132), TLR signaling was critical for this B-1 cell response, as B-1 cells in mice lacking all TLR signaling due to a lack of TLR2, 4, and Unc93 were largely unresponsive to infection (111). Mechanistically, TLR signaling caused the reorganization of the BCR complex on the B-1 cell surface, which included a loss of expression of CD5 and alterations in the signaling ability of the BCR, resulting in increased interaction between CD79a (Igα) and Syk, increased expression in the early marker of BCR activation Nur77, and increased phosphorylation of downstream BCR signaling molecule Akt (111). An important question to be resolved is the role of BCR signaling and thus the antigen specificity of B-1 cells in the regulation of their responses to infection.
Innate, Type I IFN-Dependent Activation of Follicular B Cells Primes the Adaptive Antiviral Response
The rapid innate-signaling induced activation of B-1 cells following influenza infection in the draining medLN occurs at the same location where viral components and entire virions, along with inflammatory signals that are induced in response to virus replication and host cell damage, rapidly accumulate. The medLN is of course also the destination of migratory dendritic cells (DCs) that collect antigen in the respiratory tract to initiate B and T cell responses. Accumulating evidence suggests that follicular B cells (B-2 cells, referred to simply as B cells from hereon) also play a critical role in shaping this local response.
Early influenza infection is characterized by considerable increases in the local production of Type I IFNs, a group of cytokines inducing the antiviral state. Type I IFN stimulation of B cells leads to enhanced BCR-dependent calcium flux and increases expression of many costimulatory surface receptors (18). Interestingly, Type I IFNs alone do not induce proliferation, but do enhance BCR-mediated proliferation. Presumably, this is to enable even low affinity B cells to orchestrate an antiviral response and initiate the expansion of virus-specific clones, without the entire compartment becoming a target of virus infection itself. Within 2 days of infection B cells within the medLN receive an IFN-β signal through the IFN α/β receptor (IFNAR), causing the upregulation of CD69 and the costimulatory surface molecule CD86 (26,35,66). Type I IFN-induced expression of CD69 inhibits signaling of the sphingosine-1-phosphate receptor (S1P1) (122), which controls B cell egress from LNs into the blood (75). The increased number and activation state of B cells in the follicles lead to immense changes in LN physiology that support the induction of adaptive immune responses. During Lymphocytic choriomeningitis virus (LCMV) infection, LN remodeling and increases in cellularity depend on B cell-derived lymphotoxin (LT) α-1-β-2 (65), part of a feed-forward cycle involving CXCL13 production in stromal cells, which attracts circulating B cells to the follicle through the receptor CXCR5 (9). Following immunization with keyhole limpet hemocyanin, B cell-derived vascular endothelial growth factor A (VEGF-A) was shown as critical for both LN growth and migration of DCs from the site of inoculation to the draining LN (8). However, VEGF-A was dispensable for LN expansion during LCMV infection (65), indicating that different inflammatory agents and/or antigens, while both requiring B cells, may differentially affect LN remodeling following B cell activation. Together the data indicate that B cells not only affect the local LN environment but also the migration of professional APCs from sites of primary inflammation, thereby critically regulating initiation of the adaptive response.
While Type I IFN-dependent signaling leads to the above cascade of B cell activation events during viral infection, it can also lead to early suppression of the humoral response when virally-infected B cells are eliminated (82,109) through the IFN-mediated activation of macrophages (109) and CD8 T cells (82). This was shown following infection of mice with either chronic or acute strains of LCMV, explaining the surprising finding that blockade of Type I IFN resulted in accelerated clearance of LCMV (84).
The process of virus infection-induced elimination of antigen-specific B cell clones might be more common than currently appreciated. They may be particularly permissive to viral infections, due to the internalization of virus through antigen-specific BCR-mediated endocytosis, which could provide tailor-made access for viruses into a host cell. Indeed, influenza virus has been shown to infect influenza-specific B cells in the lungs of BCR-transgenic mice, leading to B cell death (31). Early elimination of B cells may explain observations on antigen-specific antibody response kinetics: B cells home to the T:B border within hours of immunization (89), yet following influenza infection antibody secreting cell (ASC) numbers in the medLN do not rise appreciably until several days after infection (116). Thus acute viral infections can blunt antigen-specific B cell survival and expansion both directly and indirectly, potentially causing significant delays in antiviral antibody responses.
Antigen Exposure in the Follicle Is the Beginning of a Multistep Process in Determining B Cell Fate and Antibody Secreting Potential
Induction of virus-induced B cell responses requires B cell activation through binding of cognate antigens to the BCR. In the spleen and LN, B cells are confined predominantly to the follicles. Mutiple well-regulated mechanisms ensure effective exposure of naive B cells to viral antigens. The foremost of these is thought to be the delivery of antigen to follicular dendritic cells (FDCs). FDCs can acquire antigen in a multitude of ways, including directly from the subcapsular sinus (SCS) or from interfollicular sinuses (40). In addition, specialized CD169+ macrophages lining the SCS can capture antigen complexes from the afferent lymphatics using their vast array of Fc and complement receptors. Captured antigen can then be transferred to FDCs in the follicle. SCS macrophages can also present antigen directly to naive B cells leading to their activation (100).
Following subcutaneous immunization of mice with influenza, viral particles were collected both in the SCS and the medulla of the draining LN (41). While the CD169+ SCS macrophages bound virions using mannose-binding lectins, DCs in the medulla required DC-SIGN, a C-lectin receptor (41), for virion binding. Interestingly, only deletion of the medullary-resident DCs led to major decreases in ASCs in the LN and reductions in antiviral serum antibody titers, while deletion of SCS macrophages caused increased antigen dissemination but had no effect on antiviral humoral responses (41). After virus binding, DCs from the medulla rapidly moved toward the follicle, consistent with their role in initiating strong antiviral humoral immunity. The mechanism by which these viral laden medullary DCs support local ASC formation, however, remains to be more fully elucidated.
Independent of professional APCs, B cells can carry antigen directly from sites of infection to lymph tissue by both BCR-dependent (45) and BCR-independent mechanisms (14,106). Virus-like particle (VLP) laden B cells activated splenic B-cells in the absence of other professional antigen presenting cells in vitro, and increased IgG responses were seen in vivo following adoptive transfer of these cells. Thus the rapid activation of B cells following viral infection occurs through multiple pathways that ensure effective deliverance of antigen.
BCR-mediated antigen binding induces dynamic changes to their expression of chemokine receptors, resulting in the localization of antigen-stimulated B cells to specific lymph tissue subcompartments for further activation, proliferation, and differentiation. Within hours of immunization, B cells downregulate CXCR5, which retains resting B cells within the follicle, and upregulate CCR7, the receptor for CCL19/21, produced in the paracortex, leading to their migration toward the T cell zone. After migration to the T:B border, activated B cells also secrete T cell chemoattractants CCL3 and CCL4, increasing the efficiency of interaction with activated CCR5+ CD4 T cells that have also migrated to the T:B border (27). Along with regulation of CXCR5 and CCR7, the receptor Ebi2 regulates B cell positioning within the follicle. High EBi2 expression drives B cells toward the outer edge, near the SCS, and repression drives B cells toward the center, the latter contributing toward formation and sustainment of germinal centers (GCs) (99). Recently, CCR6, a receptor for macrophage-produced CCL20 (MIP3-α) (102), has been implicated in attracting and retaining memory B (Bmem) cells at the SCS (82) and multiple B cell subsets within the subepithelial dome of Peyer's patches (16). CCR6 is also expressed on naive B cells (64), but has been shown to be more responsive to chemokines after B cell activation (71). The signal may facilitate increased interactions of B cells with SCS macrophages for antigen or innate signals, leading to optimal humoral responses. The effects this chemokine axis has on B cell immunity require more in-depth investigations.
Once at the T:B border, activated B cells undergo sustained interaction with CD4 T cells through peptide-MHCII (pMHCII):TCR engagement, allowing for coreceptor activation. Stimulation of the coreceptors CD40 on B cells and its ligand CD40L on T cells is the major driver of T-dependent B cell activation. CD40 signaling leads to expression of inducible T cell costimulator ligand (ICOS-L), which engages ICOS on T cells, further strengthening the interaction around the pMHCII:TCR synapse and increasing reciprocal T and B cell activation (73,95). Formation of the immunologic synapse between B and T cells is enhanced further by lymphocyte function-associated factor 1/intercellular adhesion molecule 1 interactions (21,83,110). Simultaneously, CD40 activates canonical and noncanonical forms of nuclear factor κB (NFκB) (29), leading to increased B cell survival. Of particular interest are the effects of noncanonical NFκB on enhancing expression of the transcription factor interferon regulated factor 4 (IRF4), which is initially induced upon BCR stimulation (42) and is required for both ASC formation (115) and for sustained GC responses (135).
The Extrafollicular Response Generates the Early Antigen-Specific Antibody Response During Viral Infection
Extrafollicular (EF) B cell and GC responses are spatially, functionally, and temporally distinct outcomes of B cell activation following viral infections (Fig. 1). The EF response is characterized as a short-lived, antibody-generating response locally within secondary lymphoid tissues. They arise from the initial clonal expansion of antigen-specific B cells, which has recently been shown to give rise predominantly to nonswitched IgM memory cells (134). EF responses begin within days of influenza infection and resolve as the virus is cleared. This contrasts with GC responses, which begin to form toward the end of acute infection and then last for months. During influenza infection, the CXCR4-expressing plasmablasts and plasma cells from the EF response migrate to the CXCL12-producing fibroblastic reticular cells in the medullary cords of the medLN (48).

Generation of the adaptive B cell response in the draining LN.
The EF response has been considered to generate predominantly low-affinity antibodies. This conclusion is based primarily on immunization studies of mice with the hapten (4-hydroxy-3-nitrophenyl)acetyl (NP), whose early antibody responses have been shown to derive from early, germline-encoded antibodies of low affinity, which after seeding GC heavily mutate to acquire higher affinity for NP (35).
Viral infection models, along with more recent observations on ASC differentiation, however, have provided information on the nature of EF-derived antibodies that require a more nuanced view of their quality. For example, during infection of mice with vesicular stomatitis virus, overall avidity of serum IgG to viral antigens showed no significant increases over the course of infection (107), indicating that GCs did not enhance the overall avidity of virus-IgG over that produced by early EF responses. The levels of an early and dominant influenza hemagglutinin (HA) H1-specific IgG, encoded by the C12 idiotype in BALB/c mice, not only correlated with protection against acute viral infection but also was of high affinity, despite being encoded from germline. These C12Id-encoded responses were only present during primary but not challenge infections, that is, they lacked memory formation, and C12Id-expressing cells were not found in the GC (59,108), suggesting their origins from EF rather than GC responses. Thus, the affinity of a given antibody response primarily depends on the presence of the preinfection B cell repertoire, rather than necessarily requiring the formation of GCs.
Further supporting this conclusion are studies using transgenic mice expressing BCR specific for the model antigen hen-egg lysozyme (HEL). High-affinity interactions between HEL and BCR polarized B cells toward an EF fate, while lower affinity interactions, through amino acid substitutions in HEL, led to decreased ASC formation (98). High-affinity BCR interactions were associated with higher expression of IRF4, a transcription factor that drives expression of genes known to regulate plasma cells, such as Blimp1 and XBP1 (137). While formation of both EF and GCs require IRF4 expression, GC B cells express relatively lower amounts of IRF4 and higher amounts of the antagonizing transcription factor IRF8 (137).
These data demonstrated that early antibodies generated by EF responses during virus infection can be both, antigen specific and protective. Indeed, early antibody responses have been linked to better health outcomes during EBOV infections (69). Development of early ASCs in the EF response requires high IRF4 induction, which is achieved through high-affinity interactions between antigen and the BCR. Given that not all repertoires may contain high-affinity BCR-expressing B cells in the preinfection repertoire, other stimuli may act in concert with the BCR to induce the high levels of IRF4 expression required for plasma cell differentiation. Further studies are needed to define such signals and their integration with BCR-mediated B cell activation.
BCR-Independent Regulation of EF and GC Responses Following Virus Infections
The mechanisms behind the expansion and contraction of EF responses are also not fully understood, but at least three cell types have been linked to EF responses through production of the cytokines B cell activating factor (BAFF) and A Proliferation Induced Ligand (APRIL), the latter supporting bone marrow plasma cell survival (96,103,139): Neutrophils, which helped B cells and supported ASC formation and maintenance (49,96,103), and both, fibroblastic reticular cells and CD11c+ DCs within the LN medulla, which have been found in tight association with plasma cells (139). Determining whether BAFF and/or APRIL or individual cell types generating these cytokines are required for supporting the EF responses during viral infections is challenging, given that B cell development and survival require their presence. Numerous other cytokines have also been associated with ASC differentiation [reviewed in ref. (80)].
Interleukin (IL)-21 is the most prominent of these and its presence is necessary for optimal humoral responses through the generation and expansion of early (EF) and late (GC) plasma cells (141). The lack of CD4-derived IL-21 after influenza immunization severely crippled the antiviral antibody responses (79). IL-21 production is associated primarily with T follicular helper (Tfh) cells (19). Interestingly, Tfh cells only produced IL-21 transiently in GC after infection with the helminth Nippostrongylus brasiliensis (133). Following influenza immunization, the majority of IL-21 producers in the spleen was found not to be Tfh cells but rather CXCR3+ T-helper 1 (Th1) cells that also produced IFN-γ (79). This indicates that while IL-21 may be produced by Tfh cells that modulate GC responses, Th1 cells can also act as drivers of B cell differentiation after virus inoculation. Although this remains to be tested, it may support the generation of plasmablasts in EF responses through the interaction of local effector T and B cells. In support, sera from mice that lacked GC responses and Tfh could still generate protective IgG2 antibody responses, which were generated with help of IFN-γ and IL-21 producing Th1 effectors, while lack of GC and IFN-γ production failed to protect against influenza challenge (79), suggesting that ASCs formed outside the GC do so in concert with generation of a local, anti-viral T cell response.
GC development follows the peak of the EF responses and its development is orchestrated also by a number of different cytokines. B cells that lacked STAT6, the primary signal transducer for the IL-4/13 receptor, were unable to downregulate Ebi2, which as mentioned above regulates the positioning of activated B cells in the inner follicle for GC formation (128). Interestingly, production of IL-4 by natural killer T (NKT) cells after influenza infection is required for optimal GC responses, even when Tfhs become the predominant IL-4 producers at nine days postinfluenza infection (39). Given that NKT-produced IL-4 is present before GC formation takes place, it likely contributes toward polarization of activated B cells in seeding of GCs. Indeed, global IL-4/IL13-deficient mice had compromised GC reactions after LCMV infection (128), while Tfh-specific knockout of these cytokines had no effect. The data suggest that early production of IL-4 supports GC responses from outside the follicle.
Lack of IL-4 signaling was also shown to reduce the number of IgG1-generating ASCs in the medLN at nine days postinfluenza infection (39), which coincides with the peak of the EF response and nascent GC development. However, this work did not assess whether the lack of IL-4 affected class-switch recombination to IgG1 alone or more broadly also reduced production of IFN-mediated IgG2a and IgG2c in BALB/c and C57BL/6 mice, respectively. In addition, IL-4 has been shown to directly suppress Type I IFN signaling in DCs (124), as Signal transducers and activators (STATs) associated with IL-4/13R activation compete with STATs from IFNAR. Therefore, as IL-4 production follows production of Type I IFN in draining LNs, it may impact B cell fate decisions toward an EF or GC response. With the presence of IFN-γ necessary to form antibody responses in the absence of GC formation during influenza infection (79), shifts in IL-4 and IFN-γ production appear integral in generating early and late antibody responses, respectively. Along with IL-21, these cytokines shape the dichotomy of EF and GC responses in both a spatial and temporal manner (Fig. 2).
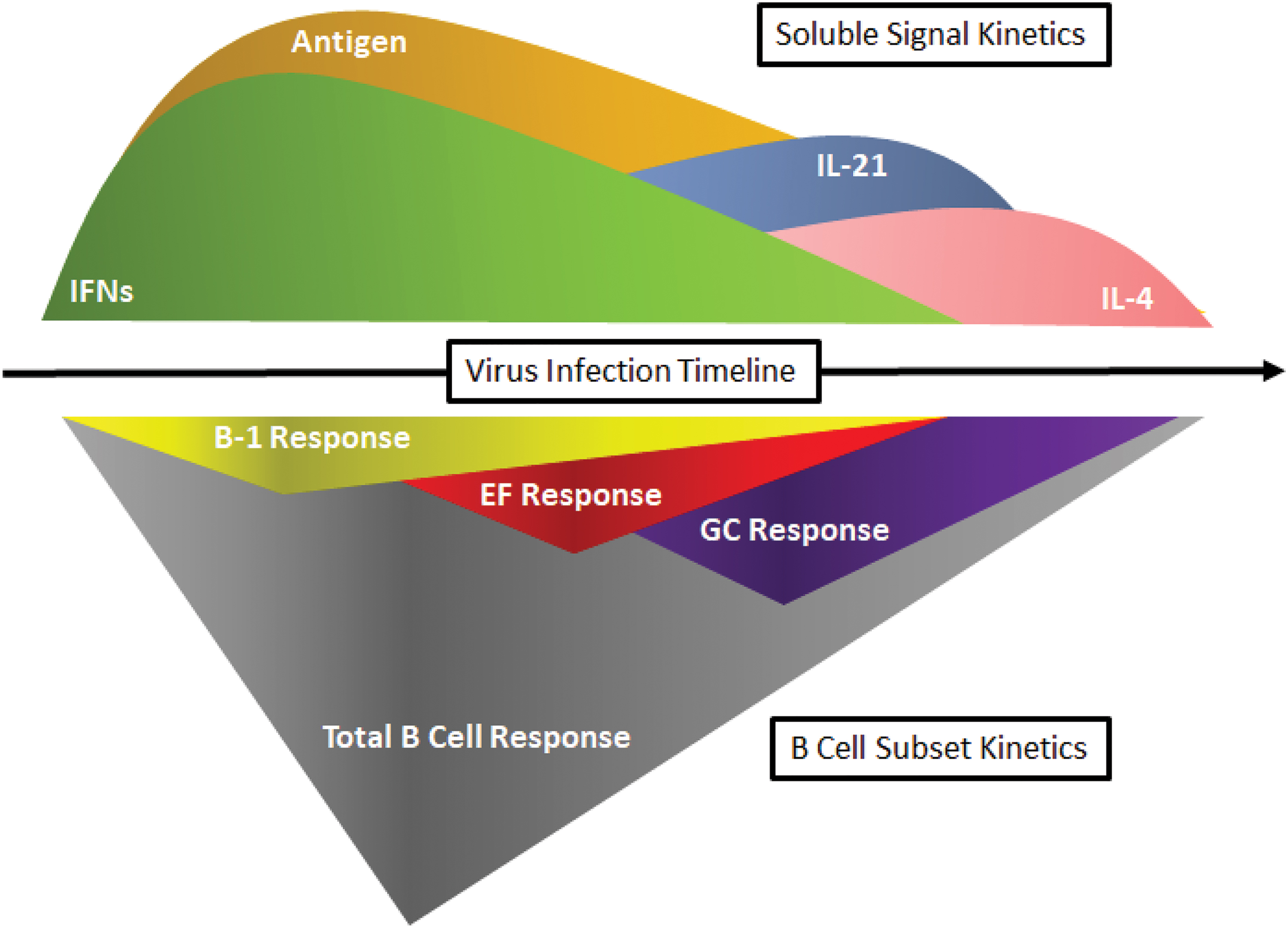
Relationship between changes in inflammatory signal kinetics in draining LN after viral infection and its effect on the B cell response. Soluble signals (top) change concentrations over the course of an acute viral infection. As the virus replicates and increases its exposure to the immune system as soluble and cell-bound antigen, the IFN response is initiated, recruiting and differentiating B-1 cells while activating B-2 cells within the draining LN (bottom), leading to their accumulation, survival, and priming them for further activation. As T cells become activated, they begin to secrete IL-21 (top), leading to B cell differentiation into plasmablasts for the EF response and contribution toward the GC reaction (bottom) later on as T follicular helper cells. IL-4 production (top) outside the follicle from CD4 T cells and natural killer T cells help initiate the GC response, but is not required for GC maintenance. As viral antigen concentrations and subsequently IFN signaling contract, so does the B-1 and EF responses, along with the total B cell population. Without competing early inflammatory signals, the GC response becomes the dominant response, lasting for extended periods of time well after virus clearance. IL, interleukin; IFN, interferon. Color images are available online.
TLR activation also contributes toward B cell activation and differentiation into ASCs, with B cell-intrinsic TLR signaling necessary for generating antiviral antibodies after immunization (58). Stimulation of TLR-4 with lipopolysaccharide strongly increased the expression of IRF4 (42), and TLR-9 signaling synergized with CD40 signaling to increase immunoglobulin class switch recombination and antibody production (94). In vitro, TLR-9 stimulation of naive B cells led to significant increased expression of prdm-1 (encoding Blimp-1) and associated ASC genes, while in vivo, it repressed antigen uptake and processing (3). This indicates that TLR-9 stimulation alone enhances terminal differentiation of B cells into ASCs, but prevents naive B cells from reaching the necessary precursor stages that precede ASC differentiation. However, different TLRs may have different effects on B cell fate decisions. Indeed, mice lacking TLR-7 had reduced frequencies of splenic GC B cells following intranasal influenza infection without reductions in influenza-specific ASCs (53). This is intriguing, as TLR-4, TLR-7, and TLR-9 all signal through the adaptor protein Myeloid differentiation factor 88, but only TLR-7 appears to support GC function, indicating that the context of additional signals is important in TLRs' determination of B cell fates.
GC Responses Generate Memory B Cells and Plasma Cells That Provide Protection from Repeat Viral Challenge
To initiate a GC response, B cells travel from the T:B border back to the follicle, dragging their cognate T cell along in a Signaling Lymphocytic Activation Molecule-associated protein (SAP)-dependent manner, without which GC formation is strongly diminished (20). In the follicle, nascent GCs expand within the FDC network of the inner follicle, eventually maturing to contain microstructures known as light zone (LZ) and dark zone (DZ). The DZ faces toward the paracortex and consists of rapidly dividing B cells known as centroblasts that undergo several rounds of somatic hypermutation (SHM) that alter the variable region of the BCR for altered interactions with antigen. To test the outcome of BCR mutations, mutated B cells leave the DZ and enter the LZ to interact with Tfh cells and antigen tethered on FDCs. B cells here are known as centrocytes and either die, return to the DZ for further rounds of SHM, or they leave the GC to become either memory B (Bmem) cells or long-lived plasma cells (129).
Existing data suggest that Bmem cells are produced early during the GC response (134). Bmem cells closely resemble naive B cells in their transcriptional profile, except for expression of often class-switched BCR, a small selection of activation markers (134) and half-lives that extend beyond the life of the mouse (55). Following influenza virus infection Bmem cells broadly distribute into blood, lungs, medLN, and nasopharyngeal associated lymphoid tissue, but relatively few disseminate into the bone marrow (57,126). Bmem cells localize to the subcapsular space of the LN through their expression of CCR6 (33,81), priming them for rapid interactions with antigen-bearing SCS macrophages and differentiation to EF plasmablast responses.
In mice, lung resident, influenza-specific Bmem cells developed by 15 days postinfection and differentiated into ASCs upon influenza re-exposure, where they conferred protection through secretion of both IgG and IgA (4,91). Cross-reactive, influenza-specific Bmem cells were found enriched in mouse lungs, compared to spleen, 20 days postinfection, with evidence that they originated locally from lung-derived GCs (2). As cross-reactive repertoires against influenza have been extensively identified in human Bmem cells (38,77), and given that the Bmem cells from West Nile Virus infection also displayed a proclivity for cross-reactivity (104), it is likely that Bmem cell generation has evolved to combat rapidly mutating viruses, sacrificing explicit high affinity and potential neutralizing capabilities for a broad recognition of many similar viral epitopes. Indeed, vaccination with H7N9 influenza in naive adults led to only a modest response of Bmem cells to conserved epitopes, but sustained expansion and affinity maturation of Bmem cells to novel epitopes (5). Following viral challenge after immunization with VLPs, Bmem cells rapidly differentiated into short-lived plasma cells (SLPCs) rather than long-lived plasma cells (LLPCs) (63), suggesting that LLPCs emerge directly from precursors in GC. Bmem cells in tertiary tissue sites can provide rapid local protection and contribute toward initiation of a more specific adaptive response by promoting formation of immune complexes.
As GCs age, they seem more likely to produce plasma cells (134), which eventually home to the bone marrow where they reside for extended periods of time as a heterogenous population of SLPCs and LLPCs. It is thought that B cell development toward Bmem and plasma cells is based on BCR affinity for their cognate antigen, with Bmem cell development requiring lower BCR affinity for antigen compared to plasma cells (101). LLPCs in the bone marrow typically produce IgG class-switched antibodies throughout the life of the host, while LLPCs at mucosal sites are IgA class switched (51,56). Because LLPCs are terminally differentiated they are unable to adapt to mutations of a future infecting virus with altered antigen binding. For highly mutating viruses like influenza, this means that LLPC-derived antibodies, and therefore circulating antibodies, may have limited cross-reactivity to newly emerging viral strains (67).
It is unknown how bone marrow survival niches for LLPCs are prioritized and if repeated viral infections lead to changes in the LLPC populations and consequently the secreted antibody repertoire. Based on the model of original antigenic sin, memory B cells from previous viral strains or mutants may detract from establishment of antibodies against novel epitopes. While there is some evidence of this phenomenon (60), elegant work using influenza with mutated or blocked HA epitopes demonstrated that the greatest increases in antibody responses were targeted toward novel antigens (7). Furthermore, immunization with different influenza mutants saw similar increases in antibody titers against other HA epitopes (7). Finally, observations made from human responses to yearly influenza vaccines demonstrated that immunizations with novel strains led to the greatest increases in both antigen-specific circulating plasmablasts and serum antibody titers (6). Thus, the actual impact the phenomenon of “antigenic sin” has on immune protection remains to be fully evaluated.
B Cells Act as Multifunctional Effectors During Viral Infections
B cells can contribute to antiviral defense also through production of a variety of inflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-1, IL-6, and IL-10, which not only act as innate signals but also influence T cell polarization [reviewed in Shen and Fillatreau 2015 (118)]. B cell cytokine production was first demonstrated in an Epstein–Barr Virus transformed human B cell line that produced IL-1 (114). After virus-induced transformation of murine B cells these cells secreted IL-5 (97). Subsequently, B cells have been identified to produce a variety of other cytokines such as TNF-α, LT, IL-6, and IL-10. During influenza infection, IL-10 production by B cells has been shown to modulate the inflammatory milieu in the lungs, buffering against the tissue damaging effects of cytotoxic T cells (138). Specifically plasma cells have been identified as strong producers of these regulatory cytokines, which is explained by their expression of IRF4 and BLIMP-1, transcription factors that regulate these cytokines (105,119).
Functions and Consequences of Antibodies During Acute Viral Infection
When B cells fully differentiate into ASCs, their secreted antibodies bind to free virion or membrane-bound viral antigen, leading to either complete neutralization of the virus or promotion of antiviral effector functions. Antibodies produced during a viral infection can include those with specificities for most viral proteins. However, the magnitude and kinetics of individual antibodies to specific viral proteins can vary significantly (116), although the mechanisms regulating these differences are unknown. Antibodies can prevent the fusion of virus with the endosomal membrane, thus preventing entry of the virus into the cytosol (17,92). Antibodies against influenza were shown to prevent the release through budding of new virus into the extracellular space by crosslinking HA (17,127) and also by preventing the cleavage of HA in newly formed viruses, which is necessary for infection of new cells (17,125). IgA may play a special role in intracytosolic neutralization of influenza virus given that it is actively transported through epithelia cells, where it can come in contact with intracellular virions (62,76).
Virus neutralization is not the only important mechanism by which antibodies contribute to immunity. Antibodies may also contribute through antibody-dependent cellular cytotoxicity (ADCC) by NK cells or macrophages. For instance, antibodies specific for the stalk region of influenza HA, a constant region among heterologous influenza strains, do not neutralize but instead promote ADCC through the binding of immune complexes to Fc receptors (30,68). In a model of EBOV infection in mice, antibody-mediated protection correlated with ADCC activity rather than in vitro neutralization (74). As ADCC leads to cell death and subsequent release of associated inflammatory signals, the cytokine milieu generated may enhance the antiviral activities of both innate subsets like NK cells or accelerate the maturation of the adaptive response.
In some instances, virus-induced antibody responses can result in antibody dependent enhancement (ADE) of infection upon secondary challenge. In this study, instead of aiding in the clearance of the virus, the antibodies increase cellular uptake of virions and thus infection, likely through Fc-receptor mediated virus uptake (11,72). This phenomenon appears to occur mostly when the challenge virus is a heterologous virus, as first demonstrated with Dengue virus (43). In fact, preexisting antibodies to Dengue or Zika virus may in some cases induce ADE to a variety of other flaviviruses (90). ADE following immunization and subsequent live virus challenge has also been suggested to occur to influenza, based on studies in pigs (130) and ferrets (61,122). ADE provides a significant potential risk to widespread vaccinations in Dengue-endemic areas in which various strains of Dengue virus and other flaviviruses circulate. A better understanding of the balance between the immune protective and the potential immune-enhancing effects of antibodies is a critical roadblock for the development of safe and effective vaccines to these pathogens.
Conclusion
During many viral infections B cells are integral to an effective immune response, both during primary infection, as well as following a secondary challenge. B cell responses are involved at all stages of the immune response, where they can control initial viral replication to limit inflammation and tissue destruction, while later responses can help eliminate the virus and protect the host from reinfections. Circulating natAbs and local B-1 cell responses dampen an initial infection and facilitate more effective adaptive antiviral IgG responses from conventional B cells. Early control of virus infection and initiation of viral clearance coincide with the rapid generation of antibodies by high affinity B cell clones during the EF response. And finally, B cell responses in the GC result in the generation of plasma cells producing a diverse repertoire of high affinity antibodies to the primary virus and Bmem cells, which can respond rapidly at sites of infection to reinfection and appear to provide antibodies with a higher potential for cross-reactivity that may be involved in secondary infection. Together these responses protect the host or contribute to protection from severe disease following reinfection. Further investigation into the mechanisms and intricacies of B cell responses to natural viral infections is needed to develop effective prophylactics that induce and harness the host immune response to viruses.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Recent work cited in this review was supported, in part, by grants from the National Institutes of Health/National Institute of Allergy and Infectious Disease R01AI117890, U19AI109962, and R01AI148652. J.H.L. received support through NIH T-32 HL007013, and F.L.S. is the recipient of a fellowship from NIH T-32 OD011147.