
Abstract
The resolution revolution of cryo-electron microscopy (cryo-EM) has made a significant impact on the structural analysis of the Pneumoviridae multifunctional RNA polymerases. In recent months, several high-resolution structures of apo RNA polymerases of Pneumoviridae, which includes the human respiratory syncytial virus (HRSV) and human metapneumovirus (HMPV), have been determined by single-particle cryo-EM. These structures illustrated high similarities and minor differences between the Pneumoviridae polymerases and revealed the potential mechanisms of the Pneumoviridae RNA synthesis.
Introduction
Resolution revolution of cryo-electron microscopy
Cryo-electron microscopy (cryo-EM) is an imaging technique for biological samples that are flash-frozen in the native environment as vitreous ice at cryogenic temperature (100 K). With recent breakthroughs of detector technology and image processing algorithms, cryo-EM leads to the resolution revolution and becomes a primary tool for the determination of macromolecular structures at near-atomic resolution, in addition to X-ray crystallography and nuclear magnetic resonance, without the need of crystals (8,38,43). The revolution is still ongoing, with increasing numbers of macromolecular assemblies being determined in different conformational states, higher resolutions being achieved, and symmetry limits being overcome (41). Collectively, cryo-EM is well suited for large heterogeneous assemblies and systems traditionally challenging for structural characterization (7,14,39).
One such challenging system is the RNA polymerase of the order of Mononegavirales, known as nonsegmented negative sense RNA viruses, which includes many significant human pathogens such as rabies virus, Ebola virus, and human respiratory syncytial virus (HRSV). There are currently eight virus families in the order of Mononegavirales, and Pneumoviridae is a new virus family since 2016 (used to be a subfamily Pneumovirinae in Paramyxoviridae) (2,30). Pneumoviridae has two genera, Orthopneumovirus and Metapneumovirus, with HRSV and human metapneumovirus (HMPV), the extensively studied representative, respectively. Both HRSV and HMPV cause severe respiratory diseases in young children, older adults, and immunocompromised patients worldwide (28,42,44). Currently, there is no effective vaccine nor antiviral therapy available to prevent or treat HRSV or HMPV infections (12,13,18,40). There are many unique aspects of RNA synthesis by the Pneumoviridae polymerases that make the polymerases attractive antiviral drug targets. Therefore, understanding the mechanisms of how Pneumoviridae polymerases function is a critical need for antiviral drug development.
RNA synthesis of Pneumoviridae
The RNA polymerase of Pneumoviridae, including that of HRSV and HMPV, is a large multifunctional polymerase complex that catalyzes three distinct enzymatic activities, namely, RNA dependent RNA polymerase (RdRp), capping (Cap), and cap methyltransferase (MT). The Pneumoviridae RNA polymerase is composed of two proteins, a large protein (L) and a tetrameric phosphoprotein (P). The Pneumoviridae L protein is a single polypeptide that contains all three catalytic domains that are necessary for RNA synthesis, cap addition, and cap methylation. The Pneumoviridae P protein is a tetramer in solution and the cofactor that regulates the activities of the L protein.
Pneumoviridae shares a common strategy of viral RNA synthesis (9,45), and it is believed to follow the “start-stop model” of sequential transcription (1,3) (features highlighted in Fig. 1). According to the “start-stop model,” (i) the polymerase recognizes a single promoter element of the leader region (3′ end of the negative-sense RNA genome) to start RNA synthesis; and (ii) the polymerase begins to synthesize messenger RNA (mRNA) in response to the first gene-start (GS) sequence (26). Immediately following transcription initiation, L caps the nascent mRNA by recognizing a specific cis-acting element at its 5′ end. The cap structure is further modified by L to become methylated at guanine-N-7 (N-7) and ribose 2′-hydroxyl (2′-O) positions. A cis-acting sequence gene end (GE) signals polyadenylation and termination; (iii) the polymerase stays on the template, reinitiates and caps the downstream mRNAs, and terminates and polyadenylates the upstream mRNAs in response to GS and GE signals of downstream genes (26,27); (iv) Like other Mononegavirales, the attenuation of the downstream mRNA occurs at each gene junction for most genes (25). Recent studies indicated the existence of nongradient and genotype-dependent transcription in RSV, suggesting the possibility of such nongradient gene expression in other Mononegavirales (34,36). Through this process of sequential attenuation of transcription, L produces ten viral mRNAs with appropriate ratios (32) (Fig. 1). Besides “start-stop” sequential transcription, L produces a leader (Le) RNA that remains uncapped and nonpolyadenylated when L initiates at the 3’ end of the genomic RNA (9,33,45). For replication, L initiates at the 3’ end of the Le or trailer complementary (TrC) sequences. However, during replication, L ignores all cis-acting regulatory signals to produce a full-length uncapped antigenome RNA instead (16). Unlike transcription, RNA replication is dependent upon ongoing protein synthesis to supply N protein to encapsidate the newly synthesized RNA (17).

The genome organization and RNA synthesis of the Pneumoviridae family. The negative-sense RSV genome is depicted 3′- > 5′ showing the leader (Le), trailer (Tr), GS, and GE regions that contain essential cis-acting signals for RNA synthesis. In Pneumoviridae, at each GS and GE, the highly conserved sequence provides critical signals for initiation and capping of downstream mRNAs and termination and polyadenylation of upstream mRNAs, respectively. The viral protein genes are shown above the mRNAs that are sequentially transcribed from them. Noncapped leader transcripts are colored in red. GE, gene end; GS, gene start; mRNAs, messenger RNAs.
Multiple cryo-EM structures of the Pneumoviridae polymerases
Due to the relatively large size of L (more than 2,000 residues in length and ∼250 kDa in molecular weight) with multiple domains and a tetrameric P that is intrinsically flexible, it is challenging to obtain crystals of the Pneumoviridae polymerase complexes for X-ray crystallography. As mentioned above, cryo-EM offers an alternative method for high-resolution structural characterization of such macromolecular complexes with limited sample quantity requirements. In recent months, there have been multiple successful reports of the structural characterizations of the Pneumoviridae polymerases by cryo-EM, two of which for HRSV (PDB: 6PZK, EMDB: EMD-20536 & PDB: 6UEN, EMDB: EMD-20754) and one for HMPV (PDB: 6U5O, EMDB: EMD-20651) (6,19,35).
The structures of the Pneumoviridae polymerases provide enriched insights into how the Pneumoviridae polymerases function. The 3.2 Å (PDB: 6PZK) and 3.67 Å (PDB: 6UEN) resolution structures of the HRSV polymerase complexes share an overall nearly identical architecture (root-mean-square deviation [RMSD] between 6PZK and 6UEN is about 1 Å) (6,19) (Fig. 2). The linear domain representation and structural organization are colored by domains (Fig. 2A). The representative raw cryo-EM micrograph, two-dimension class averages, and the three-dimension (3D) reconstruction of the HRSV polymerase (EMD-20754) are shown in Figure 2B–D. Both structures reveal that the RNA dependent RNA polymerase (RdRp, blue) and capping (Cap, green) domains of L bound with the oligomerization domain (POD, red) and C-terminal domain (PCTD, orange) of a tetramer of P. Interestingly, although both studies used full-length L and P proteins, the electron densities of MT domain and structural domains (connector domain [CD] and C-terminal domain [CTD]) of the HRSV L and the N-terminal domain of the HRSV P (PNTD) are missing in both 3D reconstructions (6,19) (Fig. 2E, missing domains are shown in gray in Fig. 2A). The integrity of the proteins was validated by mass spectrometry, and the missing electron densities suggest that those domains are disordered (6). Those disordered domains are likely intrinsically flexible, and the binding of the P tetramer to L is not sufficient to lock those domains into a stable conformation. Further comparison of both structures reveals almost identical individual domains but slightly different intermolecular arrangements, suggesting the plasticity of the L:P interface, which may adopt a more substantial rearrangement during RNA synthesis (6).
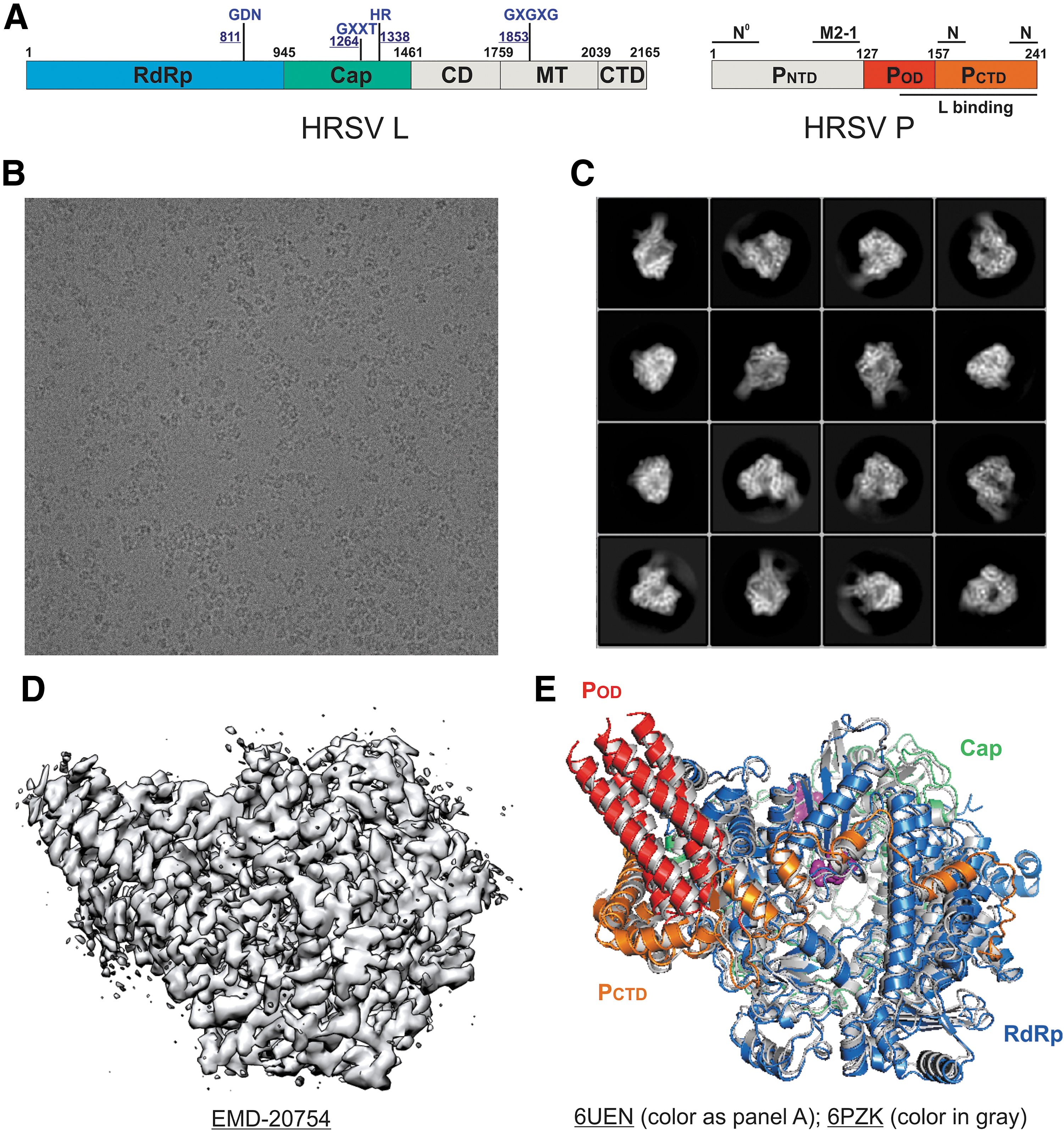
The cryo-EM structures of the HRSV polymerases.
Remarkably, the 3.7 Å resolution structure of the HMPV polymerase (PDB: 6U5O) shared a highly similar architecture to that of the HRSV polymerase that the RdRp and Cap domains of the HMPV L bind with a tetramer of the HMPV P (32). The linear domain representation and the boundaries of the HMPV L and P are shown in Figure 3A. The RMSD between HRSV and HMPV polymerase is less than 1.5 Å. Similarly, the MT and other structural domains (CD and CTD) of L and PNTD are also missing in the structure of the HMPV polymerase complex (Fig. 3B). The identities of both full-length HMPV L and P proteins used for structural characterization were also confirmed by mass spectrometry (35). Similarly, those missing domains are likely to be disordered in solution.
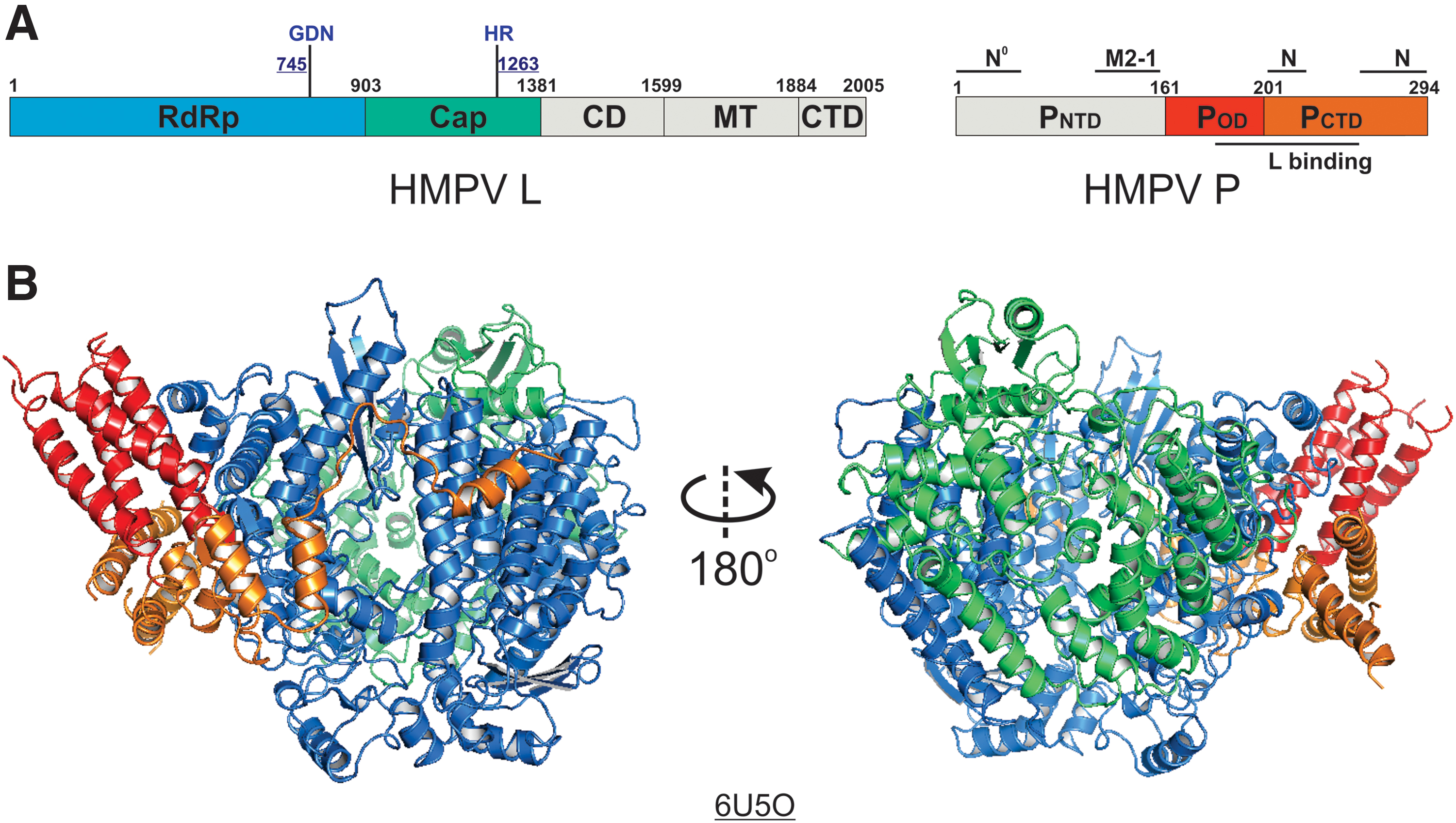
The cryo-EM structure of the HMPV polymerase.
The cryo-EM structure of a homologous Rhabdoviridae polymerase
The structure of L in complex with the N-terminal domain of P (PNTD) of vesicular stomatitis virus (VSV), a member of the Rhabdoviridae family in the order of Mononegavirales, was determined by cryo-EM at 3.8 Å resolution (PDB: 5A22, EMDB: EMD-6337) in 2015 (29). This was the first structure of Mononegavirales polymerase, requiring de novo model building of the entire L protein. Except for a few flexible loops, all three functional domains, the RdRp (blue), Cap (green), and MT (pink) domains, and two structural domains, the CD (yellow) and CTD (cyan), were resolved in the structure of the VSV L (Fig. 4). The linear domain organization is shown in Figure 4A. The RdRp domain resembles a right-hand thumb-palm-finger ring-like core domain configuration of DNA and RNA polymerases. The Cap domain of L folds next to the RdRp domain, and there was no available structural homology for the Cap domain due to the unconventional capping mechanism for Mononegavirales. Interestingly, there is a priming loop-like element in the Cap domain of the VSV L that is located next to the active site of the RdRp domain, which is speculated to function as a priming loop responsible for the de novo initiation of the RNA synthesis. The MT domain is separated from the Cap domain by the CD domain and then followed by the CTD (Fig. 4B). Due to the compact packing of the RdRp and Cap domains, the position of the priming loop-like element, and no distinct RNA product exit channel, it is speculated that the L adopts a preinitiation state, and significant rearrangements of those domains are likely to occur during other states of RNA synthesis.
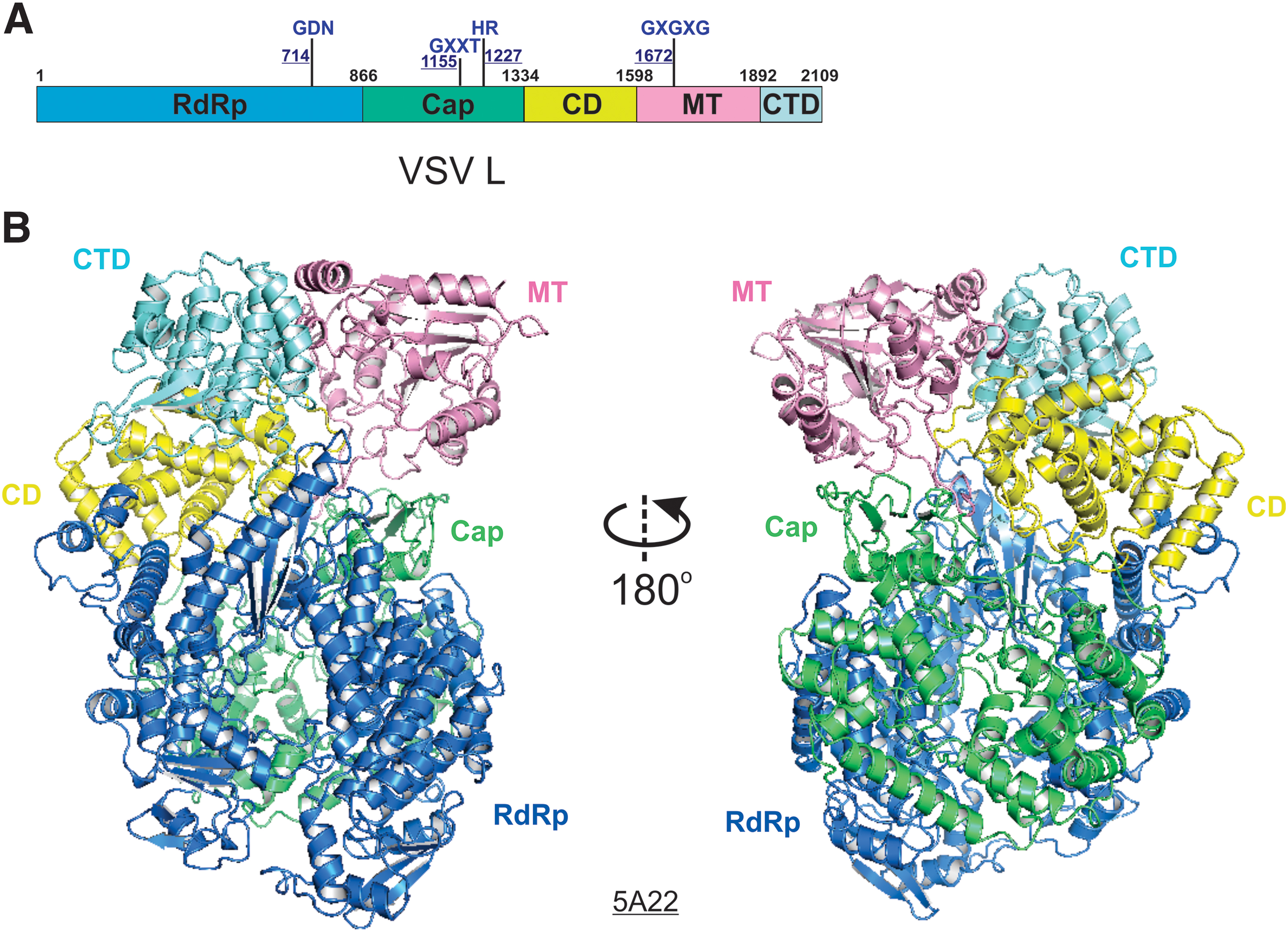
The cryo-EM structure of the VSV L.
Structural comparison of the Pneumoviridae polymerases
Recently, two high-resolution structures of the polymerases (L:P complexes) of Rhabdoviridae were reported (22,23). Given the relative conservation between the L proteins of Pneumoviridae and Rhabdoviridae (17–19% sequence identity), it was initially thought that the Pneumoviridae L might share similar overall architecture to the Rhabdoviridae L. Surprisingly, (i) only the RdRp and Cap domains of the Pneumoviridae L are visible, although both domains display similar folds to that of the Rhabdoviridae L; (ii) the priming loop-like element of the Cap domain of Pneumoviridae shows a significant shift compared to its equivalent motif of the Rhabdoviridae L, suggesting that the Pneumoviridae L is in an elongation-compatible state (Fig. 5C). Furthermore, the interactions between P and L are shown differently in Pneumoviridae and Rhabdoviridae, which may be mainly due to different domains of P that are visible in the structures, the POD and PCTD in Pneumoviridae and the PNTD in Rhabdoviridae, respectively (6,19,22,23,35). In general, the structures of the HRSV and HMPV polymerases share similar active sites GDN and HR for RdRp and Cap domains, respectively (Fig. 5B, C, highlighted in magenta spheres, Fig. 6A, highlighted in magenta text).
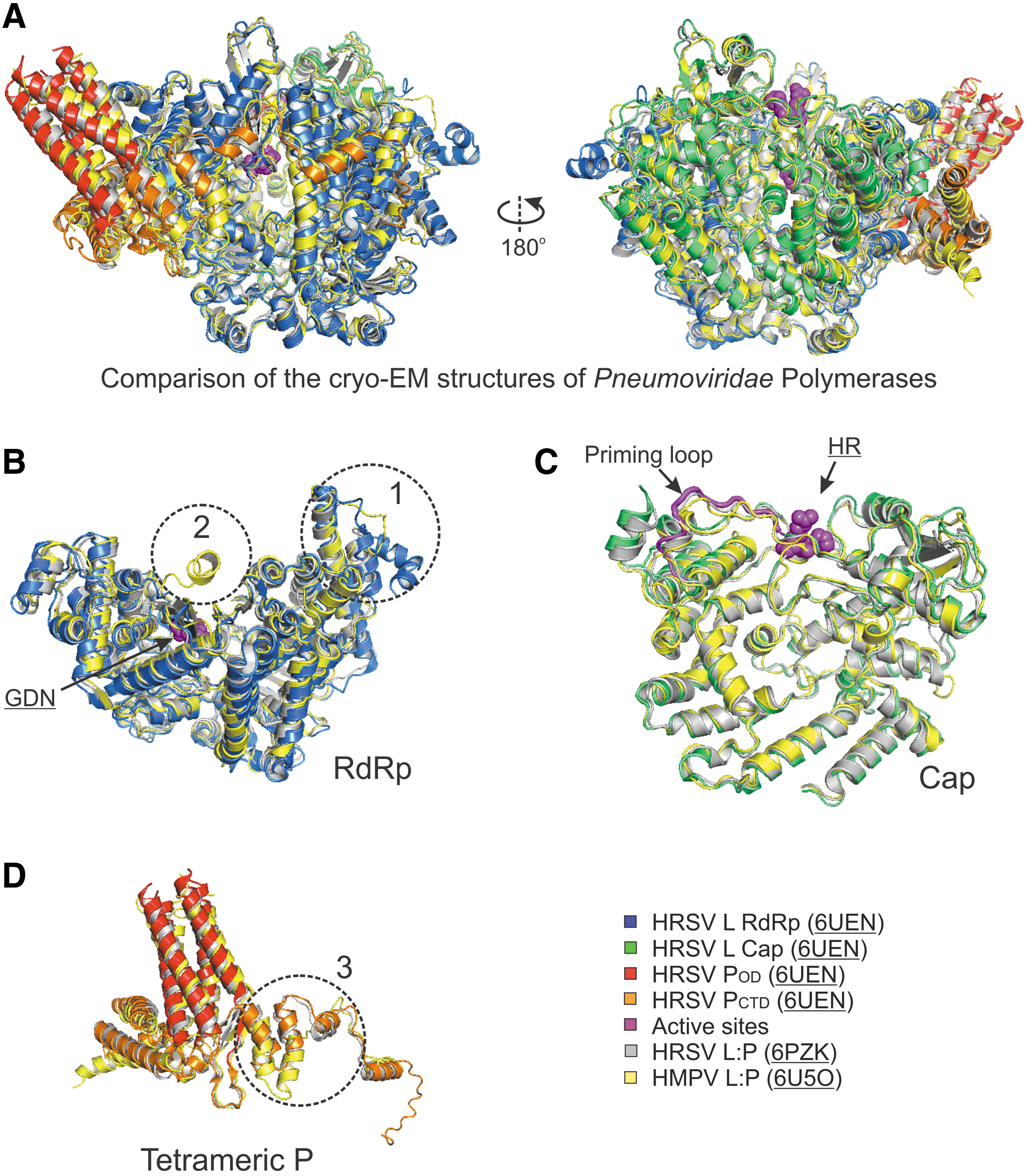
Structural comparison of the Pneumoviridae polymerases.
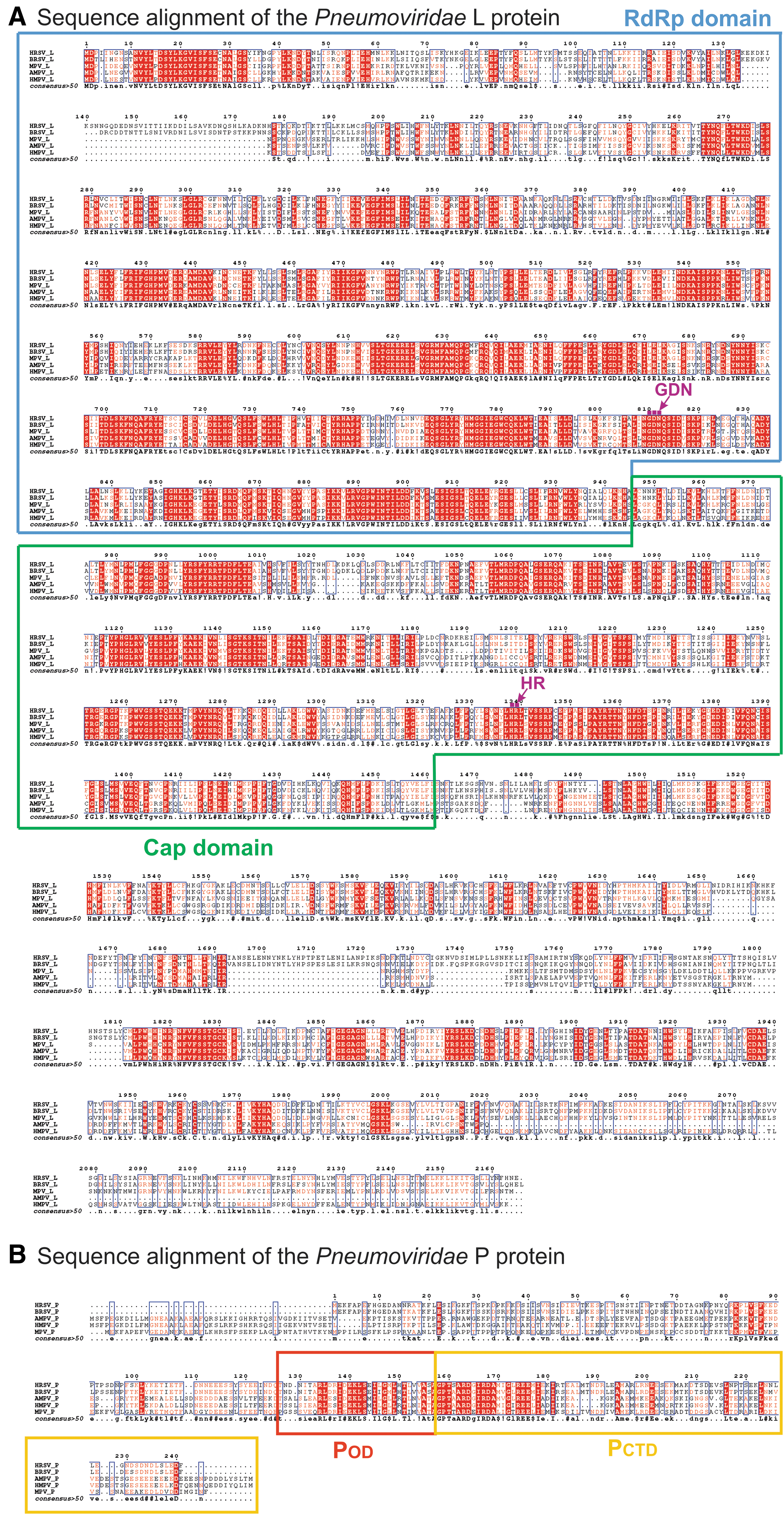
Sequence alignment of the Pneumoviridae L and P. The sequence alignment of the L proteins
Further comparison of the cryo-EM structures of HRSV and HMPV polymerases reveals that (i) the region (residues 134–176, HRSV L) in the RdRp of HRSV is mostly flexible, and some parts are not traceable, but the same region of HMPV has much shorter sequences and it is ordered (Fig. 5B, circle 1, sequence alignment in Fig. 6A), (ii) the RdRp of HRSV has a missing connecting helix (residues 660–691, HRSV L; equivalent to residues 571–597, VSV L) adjacent to the active site, providing sufficient space to accommodate RNA during RNA synthesis; however, this connecting helix can be partially modeled in the RdRp of HMPV (Fig. 5B, circle 2), (iii) the Cap domain of the HRSV L is similar to that of the HMPV L, including the priming loop-like element (magenta tubes) (Fig. 5C), and (iv) one chain of the HRSV P tetramers shows a different arrangement concerning that of the HMPV P (Fig. 5D, circle 3, sequence alignment in Fig. 6B). Those highlighted regions show subtle differences between two subfamilies, and those regions most likely contribute to the subfamily specific interactions within the RNA synthesis machine or with the host factors.
Summary
Undoubtedly, with the resolution revolution and small sample quantity requirement, cryo-EM offers an attractive solution to characterize the structural basis of challenging biological systems, such as the multifunctional Pneumoviridae polymerases. As a result, three high-resolution cryo-EM structures of the Pneumoviridae polymerases have been determined within the last few months (6,19,35). Such fast advancements reflect the power of cryo-EM as a tool for structural analysis of the Pneumoviridae polymerase complexes at different stages going forward.
As a multifunctional enzyme, the counterparts of the RdRp domain of L in eukaryotic cells are RNA polymerase (such as RNA polymerase II) (11,20), and the counterparts of the Cap and MT domains of L are RNA triphosphatase, guanylyltransferase, and MT, three of which are responsible for the addition of the methylated 5′ cap to the mRNA (10,15,21,24,31,37). Besides, L also polyadenylates at the 3′ end of the nascent mRNA, where the same process needs multiple enzymes to first cleave the 3′ end by a set of proteins (such as CPSF, CstF, and CFI) and then add the poly(A) tail by a polyadenylate polymerase (4,5,10,15,21,24,31,37). Remarkably, the same L protein ignores all transcriptional signals to replicate the entire genome during replication, without capping and polyadenylation. Therefore, it is intriguing to know how the polymerase interacts with RNA and how drugs would inhibit the functions of the polymerase. So far, there are no structures of Pneumoviridae polymerases in complex of RNA or inhibitors, and the modeling of the potential RNA interactions and druggable sites are described (6).
In summary, the sequence identities of L and P proteins between HRSV and HMPV are 48% and 36%, respectively (Fig. 6). As expected, the high structural similarity of the Pneumoviridae polymerases agrees with the high sequence conservation. Nonetheless, the structural differences also reflect the missing or insertion regions compared to each other. Collectively, those high-resolution cryo-EM structures provide insights into the molecular architectures, the interaction surfaces, the RNA synthesis mechanism, and the inhibitor developments of the Pneumoviridae polymerases.
Footnotes
Acknowledgments
The authors thank the members of the Liang lab for helpful support.
Author Disclosure Statement
The authors declare no competing interests.
Funding Information
The research programs in the B.L. laboratory are supported by the US National Institute of General Medical Sciences (NIGMS), National Institutes of Health (NIH) under award number R01GM130950, and the Research Start-Up Fund at Emory University School of Medicine.