
Abstract
Autophagy is involved in the pathogenesis of multiple pathogen infection. Previous studies have reported that human cytomegalovirus (
Introduction
Cytomegalovirus (CMV) is a widespread opportunistic pathogen virus, which can infect a variety of organs, such as the liver, lung, and nervous system. Congenital
Murine
Livingston-Rosanoff et al. (28) also found that MCMV can disrupt hepatocytes directly through viral replication. Apart from the above, suppression of immune responses induced by MCMV and interference with cytokine induction or activity also determine the severity and outcome of infection (4,43). Some studies suggested that host immune responses to MCMV infection contribute to liver immune damages through overproduction of proinflammatory cytokines, such as IFNs, proinflammatory cytokine interleukin1-β (IL-1β), and tumor necrosis factor-a (TNF-a) (28,47).
Autophagy is a highly conserved intracellular degradation pathway, which begins when the cell components are to be broken down encapsulated by a biphasic membrane creating the “autophagosomes.” The autophagosomes then fuse with the lysosomes to create “autolysosomes” and the cell components are finally degraded by lysosomal enzymes. During viral infection, autophagy can resist the infection by degrading the virus components, and induce a series of innate and adaptive immune response (14,26). Correspondingly, with long-term evolution, the viruses have developed several strategies to evade the antiviral effect of autophagy by escaping the capture of autophagic vacuoles, inhibiting the activation and maturation of autophagic vacuoles, even using some pathways of autophagy and autophagy-related proteins to promote viral replication and production (16). Published studies have shown that autophagy is involved in viral hepatitis. Both hepatitis B virus (HBV) and hepatitis C virus (HCV) have been reported to utilize autophagy for their replication, and autophagy can even mediate HBV X protein (HBx)-induced nuclear factor kappa B (NF-κB) activation and proinflammatory cytokine expression that contribute to chronic liver inflammation and fibrosis (29). Nevertheless, the report of autophagy in the modulation of MCMV hepatitis, remains undefined. As for the interplay of HCMV and autophagy, it has been demonstrated that autophagy flux was increased as early as 12 h after HCMV infection, then it was subsequently inhibited after 24 h through the interaction between viral proteins TRS1 and autophagy key signals (10,37,52). What interests us is whether autophagy is involved in virus infection-induced hepatitis and its effects on inflammatory cytokine expression, then how does autophagy function during hepatitis caused by
Materials and Methods
Animals
Four-week-old female BALB/c mice were purchased from the local experimental animal research center (Wuhan, China). Mice were maintained in a 12-h light cycle changing with a 12-h dark cycle and in the absence of specific pathogens with unlimited access to food and water. The mice of MCMV-infected group were injected with 1 × 104 PFU MCMV Smith strain in 200 μL supernatant of salivary gland homogenate after centrifugation and the mice of the mock group were injected with 200 μL Dulbecco's modified Eagle's medium (DMEM) intraperitoneally. The livers and peripheral blood were aseptically collected on days 1, 3, 7, 14, and 21 postinfection (p.i.). The autophagy intervention models were divided into six groups: uninfected group (Mock), MCMV-infected group (MCMV), starvation-treated uninfected group (Mock+S), starvation-treated MCMV-infected group (MCMV+S), chloroquine (CQ)-treated uninfected group (Mock+CQ) and CQ-treated MCMV-infected group (MCMV+CQ). Starvation was given for 48 h before harvest and freely drinking; CQ was administered at 100 mg/kg/day (dissolved in phosphate-buffered saline) for 3 days through intraperitoneal injection, the last administration time was 4 h before harvest. Mice were sacrificed on days 3 and 7, respectively, p.i. The treatment of animals in this study was confirmed and approved by the Institutional Animal Care and Use Committee of Tongji Hospital of Huazhong University of Science and Technology.
Virus and viral titer
MCMV Smith strain was obtained from salivary glands of BALB/c mice infected with MCMV and the viral titer of MCMV was determined by standard plaque assay on mouse embryonic fibroblast cell (19). The average titer of this experiment was 2.17 × 106 PFU. Aliquots of stock virus were stored at −80°C, and a fresh aliquot was thawed and diluted to the appropriate concentration for each experiment.
Western blot analysis
Proteins were extracted from frozen mouse livers and homogenized in RIPA buffer (Servicebio, China) with protease inhibitor cocktails (Servicebio). Protein concentrations were determined with the BCA Protein Assay Kit (Servicebio) and enzyme-labeled instrument. Protein samples were separated on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% skim milk in tris-buffered saline and tween 20 for 2 h at 4°C, and incubated with primary antibodies against SQSTM1/p62 (1:1000; 5114s; Cell Signaling Technology), LC3A/B (1:1000; 12741s; Cell Signaling Technology), PI3K (1:1000; 4292s; Cell Signaling Technology), mammalian target of rapamycin (mTOR) (1:1000; 2983s; Cell Signaling Technology), Akt (1:1000; 4691s; Cell Signaling Technology), p-Akt (1:1000; 4060s; Cell Signaling Technology), p-mTOR (1:1000; 5536s; Cell Signaling Technology), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:3000; 10494-1-AP; Proteintech) in primary antibody dilution buffer (Servicebio) at 4°C overnight. Membranes were incubated with secondary antibody at room temperature for 2 h and then bands were visualized by enhanced chemiluminescence. Finally, ImageJ was used to determine the expression levels of proteins.
Quantitative real-time polymerase chain reaction
Total RNA from the separated livers was extracted with TRIzol reagent (Invitrogen). The concentrations of RNA were determined using NanoDropTM 2000 Spectophotometer (Thermo Scientific). cDNA was synthesized with Hifair® II First-Strand cDNA Synthesis SuperMix (Yeasen, China) according to the protocol supplied by the manufacturer. The mRNA levels of MCMV glycoprotein B (gB), IL1-β, interferon α (IFN-α) in livers were determined with Hieff™ qPCR SYBR® (Green Master Mix Yeasen, China) on the CFX Connect™ Real-Time PCR System Bio-Rad, USA). The PCR conditions were set up for an initial 5 min denaturation at 95°C and then each cycle melted for 10 sec at 95°C, primer annealed for 20 sec at 55–60°C, extended 20 sec at 72°C. All data were normalized to GAPDH and analyzed by the 2−ΔΔCT method.
The primer sequences were as follows: mouse GAPDH, sense, TGTGTCCGTCGTGGATCTGA, and antisense, TTGCTGTTGAAGTCGCAGGAG; MCMV gB, sense, GCGACATACACTTCTCCATT, and antisense, CAGAATACGTGGCTCACA; mouse IL1-β, sense, CACTACAGGCTCCGAGATGAACAAC, and antisense, TGTCGTTGCTTGGTTCTCCTTGTAC; mouse type I IFN-α, sense, ATTTCCCCTGACCCAGGAAGATG, and antisense, CCCAGCATTGGCAGAGG.
Histopathological examination of livers and serum alanine aminotransferase and aspartate aminotransferase analysis
The assessment criteria were as follows: liver tissues were sectioned from mice of each group, fixed in 4% paraformaldehyde, and embedded in paraffin. Each sample of 4 μm section was stained with H&E. The criteria of pathological damages in liver tissues were assessed according to the Knodell liver tissue histological activity index (HAI). Three slices were selected from each group, and five high-power fields were selected randomly from each slice. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by Assay Kits (Njjcbio, China).
Immunofluorescence analysis
Paraffin sections of liver tissue were dewaxed with dimethyl-benzene and permeabilized with 0.3% Triton X-100, and 5% goat serum was used to block nonspecific binding sites. Then, liver sections were incubated with anti-light chain 3 (LC3) II (1:100; 3868; Cell Signaling Technology) and anti-LAMP1 (1:100; A16894; ABclonal) primary antibodies overnight at 4°C. The sections were further incubated with Alexa Fluor 488 and Alexa Fluor 594 secondary antibodies for 1 h at room temperature. Nuclear staining was carried out with 4,6-diamidino-2-phenylindole (DAPI) before observing with fluorescence microscope. ImageJ software was used to analyze the percentage of positively stained cells.
Immunohistochemistry analysis
Paraffin sections of liver tissue were deparaffinized for antigen retrieval. Then 5% goat serum was used to block nonspecific binding sites. Afterward, the sections were incubated with anti-LC3 II (1:200; 3868; Cell Signaling Technology) and anti-LAMP1 (1:200; A16894; ABclonal) primary antibodies overnight at 4°C. Then sections were incubated with secondary antibodies for 1 h at room temperature. All the sections were visualized using diaminobenzidine (DAB). Hepatocytes positively stained for LC3 II and LAMP 1 were examined under a light microscope at a magnification of × 200, and ImageJ software was used to analyze the positive staining intensity and the percentage of positively stained cells.
Statistical analyses
GraphPad Prism version 8 and SPSS Statistics software version 24 were used and the data were presented as the mean ± standard error of the mean. One-way analysis of variance (ANOVA) and two-way ANOVA were used for statistical analyses. p-values <0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, # p < 0.05, ## p < 0.01, ### p < 0.001, ns: not significant).
Results
Histopathological examination of liver tissues during MCMV infection
BALB/c mice were infected with 1 × 104 PFU MCMV through intraperitoneal injection to establish the MCMV hepatitis model. H&E staining of liver, HAI scores, as well as AST and ALT levels of serum samples on days 1, 3, 7, 14, and 21 p.i. were detected to evaluate the severity of liver injury. The results showed that the structure of hepatic tissue was clear and complete without significant pathological changes in control mice (Fig. 1a). In infected mice, the pathological changes aggravated gradually, a small number of inflammatory cell infiltration in hepatic lobules were observed on day 1 p.i. (Fig. 1Ab); focal inflammatory infiltration in hepatic lobules and perivascular area was observed, hepatic cords and hepatic sinus were slightly disordered while the structure of hepatic lobules was basically intact on day 3 p.i., (Fig. 1Bb); on day 7 p.i., the pathological injury reached the most severe state, the organizational structure of liver was severely damaged and significant inflammatory infiltration occurred in hepatic lobules, perivascular and portal area; focal necrosis occurred; and central venous congestion (Fig. 1Cb); this situation improved on day 14 p.i., but few focal inflammatory infiltration in hepatic lobules and portal area were still shown (Fig. 1Db); inflammatory infiltration persisted until 21 days, whereas the lobular structure had mostly returned to normal at that time (Fig. 1Eb).
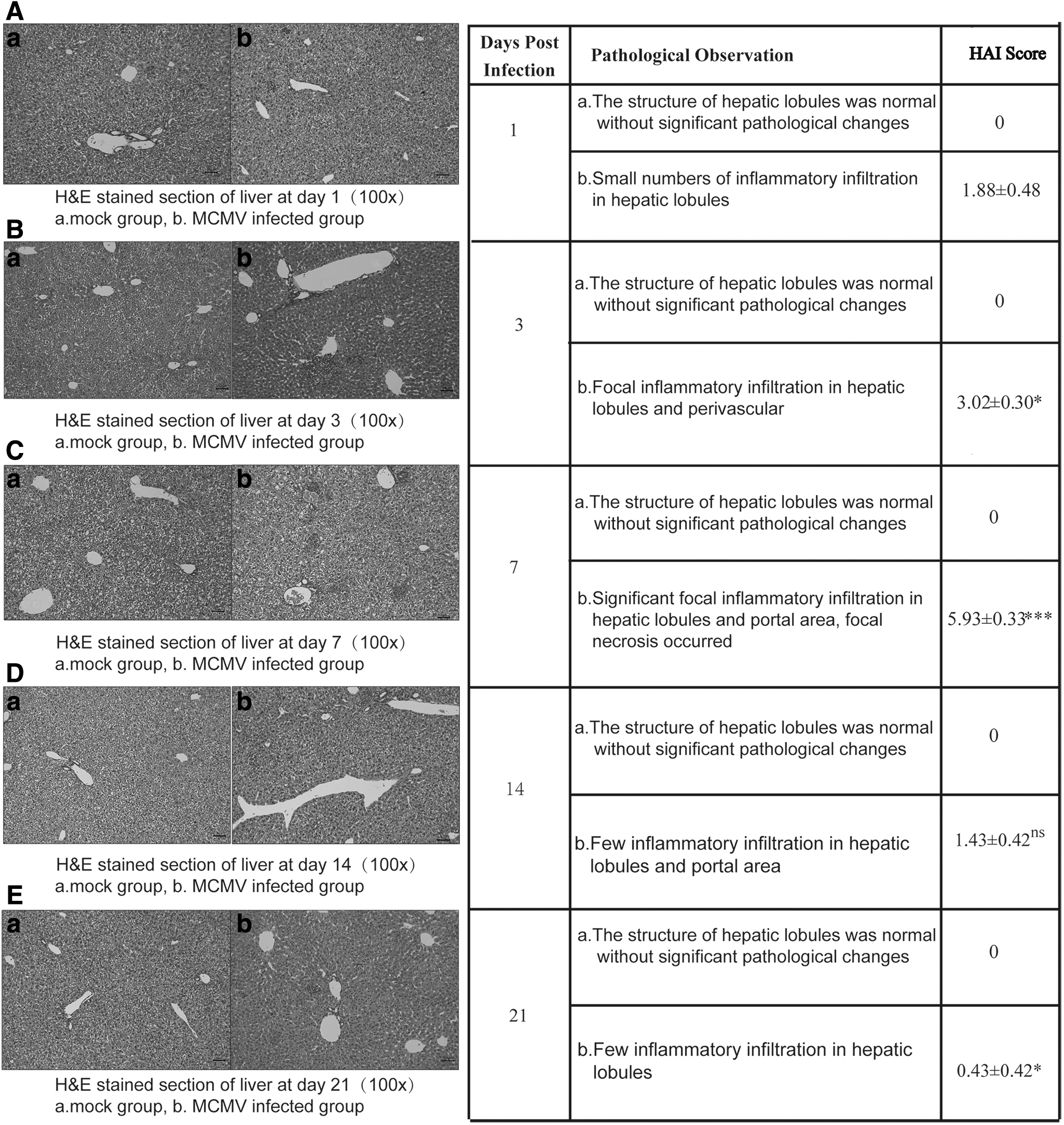
Histopathological examination of liver tissues during MCMV infection. H&E-stained liver sections of BALB/c mice at day 1 p.i.
The pathological HAI scores were consistent with the pathological changes in the livers in each infected group. In day 7-infected group, the HAI scores were significantly higher compared with day 1-infected group (p < 0.001, Fig. 2A). The activities of AST and ALT in peripheral blood, known as the liver damage markers, the results showed that the level of AST in infected mice was increased from day 1 p.i., reached the highest peak on day 7, and returned to normal level on day 14. The elevation of ALT was slightly later than AST, but also reached the highest peak on day 7 p.i. and returned to normal level on day 21. Serum AST and ALT levels in control mice had always been at a basically stable level (Fig. 2B). Considering the important role of inflammatory cytokines in liver inflammation during MCMV infection, the expression of proinflammatory cytokine IL-1β and type I IFN-α were detected using quantitative real-time polymerase chain reaction (qRT-PCR) (Fig. 2C). The expression of IL-1β and IFN-α were both increased on day 1 p.i., reached the highest on day 7, and then decreased until day 21 when it returned to normal level. The peaks of IL-1β and IFN-α production correlated with the severity of liver pathological injury (peaked on day 7 p.i.).
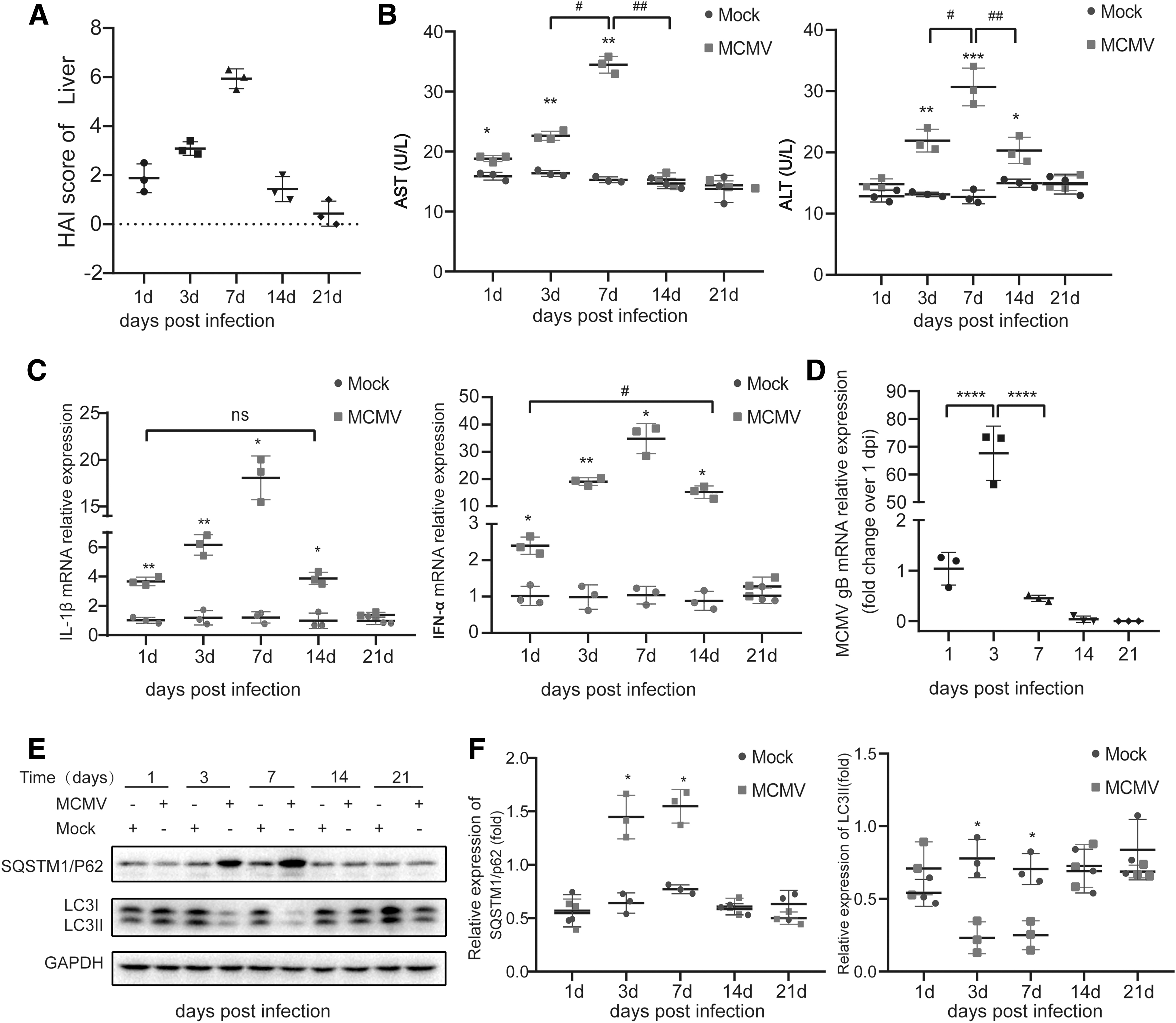
Autophagy was inhibited during MCMV hepatitis.
Transcription of MCMV gB in liver tissues during MCMV infection
The transcription level of MCMV gB was detected through qRT-PCR assay to assess MCMV viral replication in the liver (46). The transcription level of MCMV gB on day 1 p.i. were used as a benchmark. The results showed that the relative expression level of MCMV gB mRNA in infected mice reached the highest on day 3 p.i. and then decreased. On day 7 p.i., the relative expression level of MCMV gB mRNA was significantly less compared with day 3 p.i.; it could not be detected on day 21 p.i. (Fig. 2D). These results were consistent with the MCMV titers assayed through plaque method by Henson et al. (20) that MCMV peak titers occurred on days 2 to 4 p.i. and infectious virus persisted in the liver during the first 10 days after infection.
Autophagy was inhibited during MCMV hepatitis
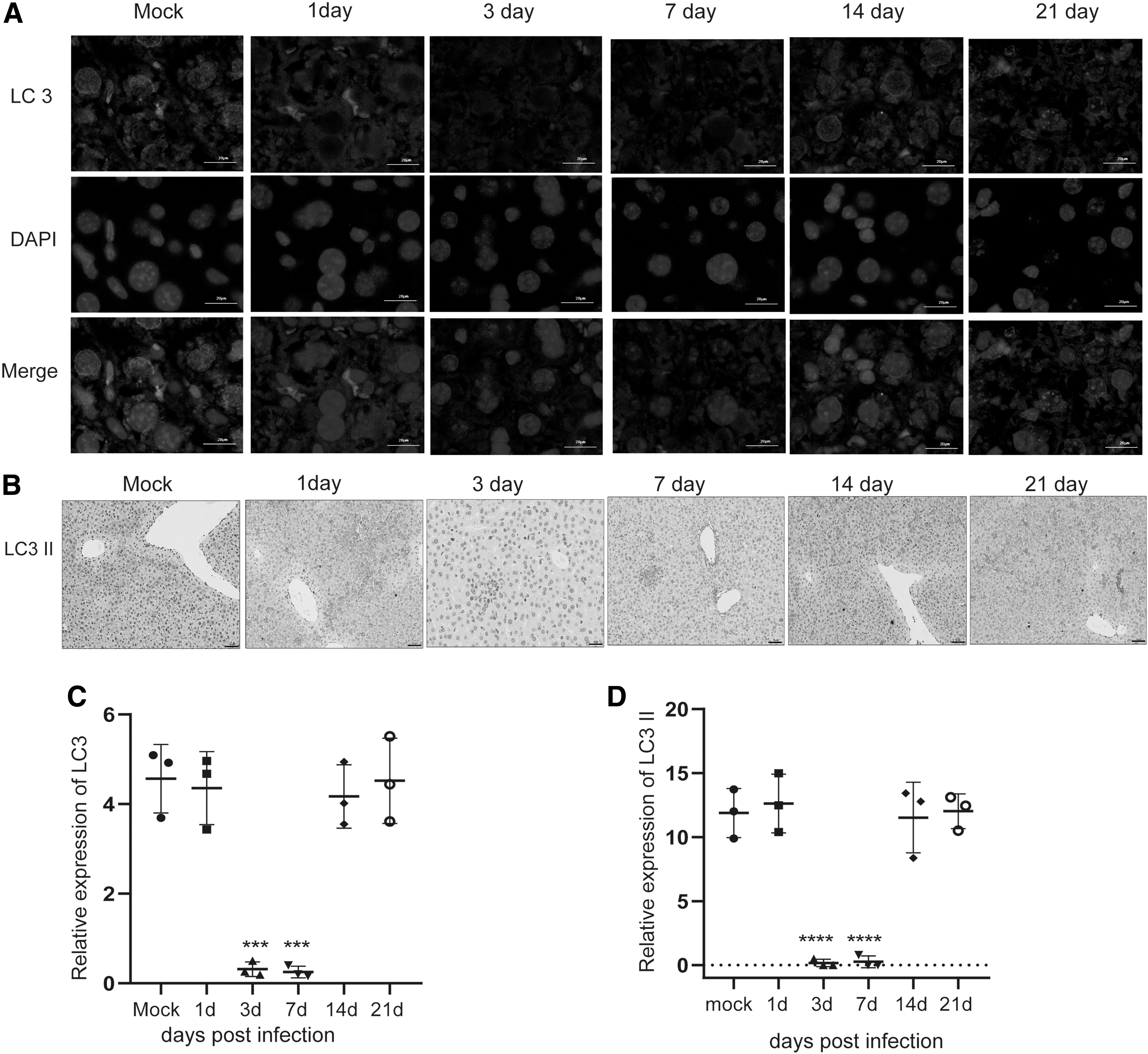
Several autophagy-associated proteins were tested to investigate autophagy level changes during MCMV infection. Microtubule-associated protein 1 LC3 is particularly relevant to autophagosome membrane and plays a key role in the amplification step of autophagosome formation. Forms of nonlipid (LC3-I) and lipid form LC3-II) are two forms of LC3. The initiation of autophagy is characterized by the lipidation of LC3-I in the membrane, LC3-I conjugates with phosphatidyl ethylamine engagement, and results in the formation of LC3-II. The expression of LC3-II is positively correlated with autophagosomes. The polyubiquitin-binding protein, sequestosome 1 (SQSTM1, also called p62), is specially degraded in autolysosomes and its expression is inversely correlated with autophagy level. Therefore, the levels of LC3-II and p62 can be widely used to monitor autophagic flux (32). In this study, we analyzed the expression levels of LC3-II and p62 proteins of the liver samples through western blot assay. Increased level of p62 and decreased level of LC3-II were observed in days 3 and 7-infected group compared with mock group, indicating that decreased autophagic activity occurred in the liver on days 3 and 7 post MCMV infection (Fig. 2E, F). However, there was no statistically significant difference in p62 and LC3-II protein expression on days 1, 14, and 21 p.i. compared with the corresponding mock group. To further determine whether autophagy was involved in MCMV infection, immunofluorescence and immunohistochemistry were performed to assess the expression of LC3-II. As shown in Figure 3A, C, the numbers of LC3 dot was decreased in days 3- and 7-infected group compared with the mock group, whereas there was no statistically significant difference in the numbers of LC3 dot on days 1-, 14-, and 21-infected group compared with mock group. The results of immunohistochemistry were consistent with that of immunofluorescence (Fig. 3B, D). These data indicated that autophagy of the liver was inhibited on days 3 and 7 post MCMV infection.
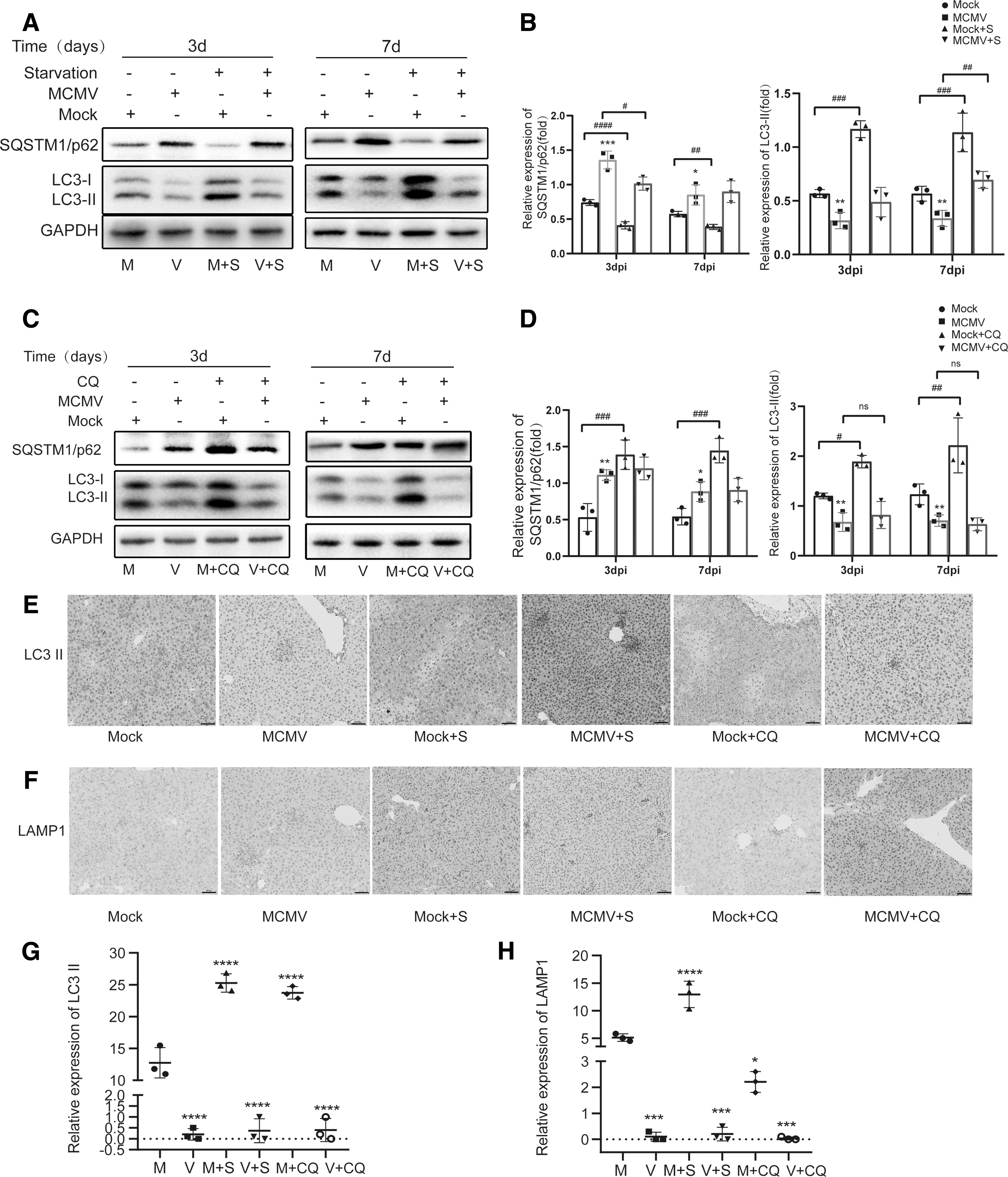
Autophagy promotes MCMV gB transcription and aggravates MCMV hepatitis
To determine how autophagy functions during MCMV hepatitis, starvation after MCMV infection was used to induce autophagy, and CQ, a negative regulator of autophagy through blocking the autophagosome/lysosome fusion step, and inducing the accumulation of LC3-II and SQSTM1/p62, was used through intraperitoneal injection to inhibit autophagy (36). The results of western blot showed that LC3-II was increased and p62 was decreased in starvation-treated uninfected group compared with untreated uninfected group both on days 3 and 7 (Fig. 4A, B). Contrarily, the intensity of LC3-II and p62 were both increased in CQ-treated uninfected group compared with untreated uninfected group on days 3 and 7 (Fig. 4C, D), suggesting that the liver of BALB/c mice is sensitive to positive and negative regulation of autophagy. In addition, immunohistochemistry was performed to assess the expressions of LC3-II and lysosomal marker LAMP1 on 3dpi. As shown in Figure 4E–H, the expression of LC3-II and LAMP1 were both increased in Mock+S group compared with the mock group, suggesting that autophagic flux was increased, whereas the expression of LC3-II was increased and the expression of LAMP1 was inhibited in Mock+CQ group compared with mock group, indicating that the fusion of autophagosomes and lysosomes was blocked. It is worth noting that the expression of LC3-II and LAMP1 were both inhibited in MCMV+CQ group compared with Mock+CQ group, and the expression of LC3-II did not have an additive increase compared with that of MCMV group. To further analyze the process of autophagy, colocalization of LC3 dots and lysosomal marker LAMP1 (LC3+LAMP1 dots) was evaluated by immunofluorescence. The number of LC3+LAMP1 dots in MCMV group, MCMV + S group, and MCMV+CQ group was significantly decreased compared with the corresponding mock group on 3dpi. (Fig. 5). These results indicated that MCMV might inhibit autophagy through blocking the early step of autophagy before the formation of autolysosomes.
Analysis of SQSTM1/p62, LC3-I, and LC3-II expression in the livers during MCMV infection.

MCMV viral replication of liver under starvation and CQ treatment was evaluated through measuring the transcription level of MCMV gB mRNA by qRT-PCR assay. Compared with MCMV group, the MCMV gB transcription level was increased in MCMV + S group on both days 3 and 7 p.i. (Fig. 6A). Contrarily, the transcription level of gB was decreased in MCMV+CQ group compared with the MCMV group on days 3 and 7 p.i. (Fig. 6C). These results suggest that enhancing autophagy promoted MCMV replication in BALB/c mice, which is consistent with previous research that stimulating autophagy could enhance HCMV viral replication (37,52).

Autophagy promotes MCMV gB transcription and aggravates MCMV hepatitis.
H&E staining and the pathological HAI score results showed that the liver pathological damages in MCMV-infected mice were more severe under starvation treatment (Fig. 7). Compared with day 3 MCMV group, the MCMV + S group demonstrated marked swelling of hepatocytes, pronounced focal hepatic necrosis associated with inflammatory cells infiltration in perivascular and portal area, hepatic portal vessel and central vein congestion, but there were still some normal liver lobular structures (Fig. 7A). In the MCMV + S group of day 7 p.i., diffuse hepatocellular swelling and necrosis seen in the liver, hepatic cords, and lobular structures had disappeared. Significant inflammatory infiltration and necrotic foci were distributed around the hepatic parenchyma and portal areas. The structure of hepatic lobules was complete in both mock group and mock+S group (Fig. 7C). The HAI scores of livers in MCMV + S group of days 3 and 7 were higher compared with the corresponding MCMV groups (Fig. 7B, D, p < 0.05). On the contrary, the pathological changes were clearly alleviated in MCMV+CQ group on day 3 p.i., less inflammatory infiltration was observed than the MCMV group, the structure of the hepatic lobules was clear, and the hepatic cords were normal (Fig. 7A). The HAI score in MCMV+CQ group was also decreased compared with the MCMV group on day 3 p.i. (Fig. 7B, p < 0.05). However, swelling of liver cell, destroyed lobular structures, and a large number of perivascular inflammatory cell infiltration were observed in both MCMV group and MCMV+CQ group on day 7 p.i., and no statistically significant difference of HAI scores was found between these two groups (Fig. 7D). The expression levels of serum AST and ALT were consistent with the results of H&E staining (Fig. 6B, D).

Histopathological examination of livers during MCMV infection treated with starvation.
We also detected the expression of inflammatory cytokines IL-1β and IFN-α using qRT-PCR assay. The expression of IL-1β in MCMV + S group was higher compared with MCMV group on both days 3 and 7 (Fig. 6E), and in MCMV+CQ group its expression was decreased compared with MCMV group (Fig. 6F). However, the expression of IFN-α in MCMV + S group was decreased compared with MCMV group on both days 3 and 7 (Fig. 6E), and in MCMV+CQ group its expression was increased compared with MCMV group on day 3, but no statistical difference was found between the two groups on day 7 p.i. (Fig. 6F). These results illustrated that inducing autophagy might somehow promote MCMV replication and aggravate MCMV hepatitis and inhibiting autophagy could suppress MCMV replication and alleviate MCMV hepatitis during the early phase of infection.
MCMV infection might regulate autophagy through PI3K/Akt/mTOR pathway during MCMV hepatitis
To further determine how autophagy was regulated by MCMV during MCMV hepatitis, the PI3K/Akt/mTOR pathway, an intracellular signaling that is involved in autophagy regulation, had attracted our attention. PI3K, Akt, mTOR, and their phosphorylation (p-Akt and p-mTOR) were examined by western blot assay.
It was observed that the expression of PI3K, the ratios of p-Akt/Akt and p-mTOR/mTOR were increased in the MCMV group on both days 3 and 7 compared with that of mock group (Fig. 8A, B), indicating that MCMV stimulated the PI3K/Akt/mTOR-signaling pathway to inhibit autophagy. In mock+S group, the expression of PI3K and the ratios of p-Akt/Akt, and p-mTOR/mTOR were decreased compared with mock group on both days 3 and 7 (Fig. 8A, B). In addition, in mock+CQ group, they were both increased compared with the mock group (Fig. 8C, D). These results suggested that starvation could inhibit the PI3K/Akt/mTOR pathway to induce autophagy, and CQ could activate this pathway to inhibit autophagy. While in the MCMV + S group, the expression of PI3K and the ratios of p-Akt/Akt, and p-mTOR/mTOR were still increased compared with mock+S group on both days 3 and 7 (Fig. 8A, B), indicating that MCMV could still manipulate autophagy by regulating other autophagy proteins to activate PI3K/Akt/mTOR-signaling pathway even in the presence of starvation.
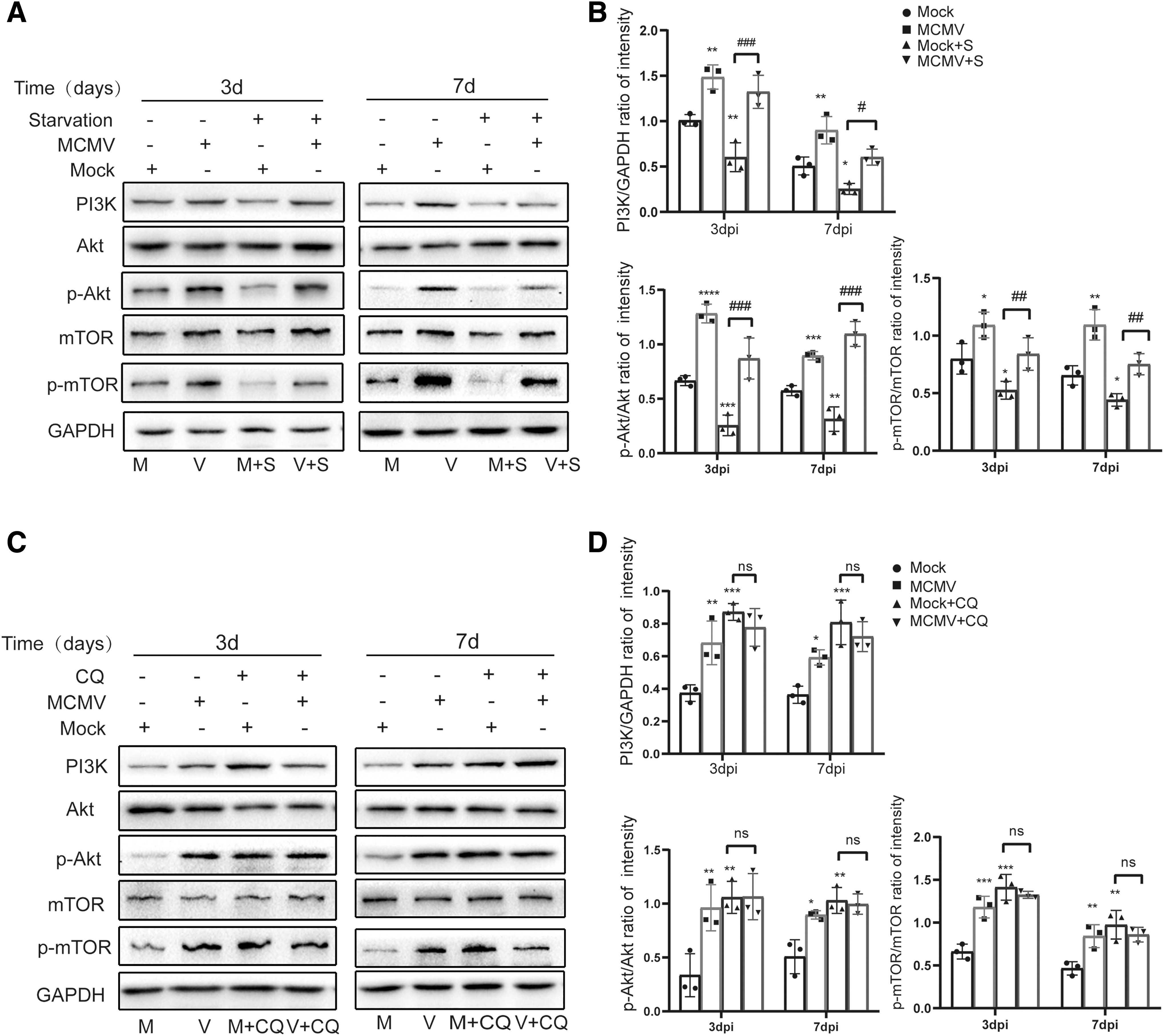
MCMV infection might regulate autophagy through PI3k/Akt/mTOR pathway.
Discussion
Recent studies have found that autophagy is closely related to virus infection, and it plays a role of “double-edged sword” in the process of virus infection. On the one hand, autophagy can eliminate intracellular pathogens by xenophagy and promote innate and adaptive immunity to participate in the control of viral infection; on the other hand, viruses have evolved strategies to escape from the host's immune response and even to utilize autophagy for their viral replication (6,13). Studies have found that autophagy is involved in the pathological process of viral hepatitis caused by HBV and HCV infection (21,29). However, it has not been reported whether autophagy is involved in the pathogenesis of
Furthermore, we also assessed the PI3K/Akt/mTOR pathway in this study. It is generally accepted that PI3K/Akt/mTOR pathway is one of the classical pathways of autophagy, the activation of mTOR complex 1 (mTORC1) can phosphorylate ULK1 and Atg13, inactivating the ULK1-Atg13-FIP200 complex and thus inhibiting the early step of the autophagy process (23). In this study, the increased expression of PI3K and the ratios of p-Akt/Akt and p-mTOR/mTOR were observed in infected mice compared with mock mice, suggesting that MCMV infection activated the PI3K/Akt/mTOR pathway. Together with the above results, we postulated that MCMV infection inhibited autophagy through blocking the early step of autophagy in mice livers. Mo et al. (35) have demonstrated that MCMV (K181 strain) infection induces autophagy in retinal pigment epithelium (RPE) cells of C57BL/6 mice at 6 h p.i. and 12 h p.i. Subsequently, MCMV inhibits autophagy by the blockade in the late step of the autophagy process after 24 h p.i., because increased expression of LC3-II, increase of GFP-LC3-positive puncta, and increased accumulation of autophagic vacuoles were observed in MCMV-infected RPE cells compared with control cells at 24 h p.i. and 3 days p.i. Moreover, xenophagy (autophagic vacuoles containing viral particle) was even observed in MCMV-infected RPE cells, which is a type of selective autophagy that involves recognition of an intracellular pathogen and targeting of the pathogen to autophagic machinery for degradation (15). In addition, they also found that MCMV infection activated mTOR signaling in RPE cells. The researchers suggested that MCMV might inhibit autophagy by blocking the late step of autophagy process, such as the formation of autolysosomes or degradation of their content, while our studies postulated that MCMV inhibited autophagy through blocking the early step of autophagy before the formation of autophagosomes. Possible reasons for these two different observations may be explained as follows. First, the use of different virus strains (MCMV K181 strain vs. MCMV Smith strain), different mice breeds (C57BL/6 mice vs. BALB/c mice), different organs (retina vs. liver) may yield significant different results. Since Chaumorcel et al. (11) demonstrated that HCMV infection inhibited autophagy in MRC5 cells through blocking the early stage of the autophagic pathway because diffuse staining of GFP-LC3, accumulation of p62, and the paucity of autophagic vacuoles were observed in infected cells. Besides, HCMV viral protein TRS1and IRS1 had been reported to inhibit the early step of autophagy by binding to autophagy protein Beclin 1 in HeLa cells (37). Those results were contrary to results reported by Mo et al. Moreover, the report by Chaumorcel et al. also noted that HCMV infection stimulated the mTOR-signaling pathway (11), which was consistent with our results. Second, the mechanisms of autophagy in vivo may be more complicated than that in in vitro because of the complexity and the variety of microenvironment in vivo situation, and the innate and adaptive immune response that may be involved in the regulation of autophagy. Third, limitations existed in our experiments. We did not provide direct evidence of autophagic activity and the presence of autophagic vacuoles at the ultrastructural level due to the lack of electron microscopy studies. The investigation of ultrastructural features of autophagy will be explored in our future experiments. Regardless, it is important to emphasize that MCMV infection could inhibit autophagy and active mTOR pathway.
Based on the question of whether autophagy affect MCMV replication, the transcription level of MCMV gB was measured by qRT-PCR technique to evaluate the effect of autophagy on MCMV replication at molecular level. Our results show that the transcription level of MCMV gB was increased in MCMV + S group compared with MCMV group, whereas in MCMV+CQ group, the expression of MCMV gB mRNA was decreased compared with the MCMV group, suggesting that autophagy was beneficial for MCMV viral replication, and it might be an important evidence and possible mechanism of autophagy aggravating MCMV hepatitis. Similarly, Mouna et al. (37) found that stimulating autophagy enhanced HCMV viral replication, conversely, and inhibiting autophagy decreased viral DNA replication in ATG16L1 knockdown cells. What is more, it has been reported that HCMV could hijack the autophagic machinery and LC3 homologs into the viral assembly compartment to promote viral replication, and autophagy-related proteins such as GABARAPL1 and GATE16 were spotted in the virions (45). From the results above, it is reasonable to believe that autophagy might be a general feature of
Multiple studies have shown that autophagy is involved in the pathogenesis of human disease and as a potent suppressor of inflammation (5,12). However, autophagy also could promote proinflammatory cytokine expression and inflammation in some biological contexts. For instance, autophagy mediates avian influenza H5N1 pseudotyped viral particles through NF-κB and p38 MAPK-signaling pathways and induce production of proinflammatory cytokines IL-1β, TNF-a, IL-6, CCL2, and CCL5 to exacerbate H5N1pp-induced lung inflammation (39). Luo et al. (29) have demonstrated that autophagy promotes the release of proinflammatory cytokines IL-6, IL-8, and CXCL2 through mediating HBx-induced NF-κB activation in viral hepatitis caused by HBV. In addition, autophagy also plays an important role for suppression of antiviral signaling during virus infection (1,44). HCV has been established to use autophagy to inhibit IFN-α production to escape host innate immune surveillance (8,9), correspondingly, inhibition of autophagy could stimulate type I IFN antiviral immunity to inhibit HCV infection (21). As with MCMV infection, it has been confirmed that large amounts of inflammatory cytokines were produced during acute hepatitis caused by MCMV infection (24,47,50). However, it has not been reported whether autophagy regulates inflammatory responses during MCMV hepatitis. In this study, we observed that the production of proinflammatory cytokine IL-1β was increased and the pathological damage of liver was more serious (included increased HAI score and elevated levels of AST and ALT) in MCMV + S group compared with MCMV group on days 3 and 7 p.i., whereas decreased production of IL-1β and alleviated pathological damage of liver (included decreased HAI score and low levels of AST and ALT) were observed in MCMV+CQ group compared with MCMV group on day 3 p.i., and these results suggested that autophagy might promote the expression of proinflammatory cytokine IL-1β to exacerbate liver pathological damage and inflammation. On the contrary, we found the production of antiviral cytokine IFN-α was not consistent with the production of IL-1β under the intervention of autophagy, the production of IFN-α was decreased in the presence of autophagy inducer starvation, whereas it was increased in the presence of autophagy inhibitors CQ compared with MCMV group. It has been reported IFN-α receptor-deficient mice (IFNAR−/−) are more susceptible to MCMV and displayed strong leukocyte infiltration and more inflammatory foci in the liver than wild type mice (3,24). Taken together with our results, it suggests that autophagy could inhibit IFN-α antiviral immune response during MCMV infection to aggravate liver inflammation. Therefore, the above observations suggested that autophagy mediates the production of inflammatory cytokines, and might be another important mechanism to participate in the pathogenesis of MCMV hepatitis. Nevertheless, its exact mechanisms require further in-depth study.
Conclusion
The present study demonstrated that autophagy in the liver of BALB/c mice is inhibited through activating the PI3K/Akt/mTOR pathway on days 3 and 7 post MCMV infection, and we hypothesized that this may be the outcome of the interaction between host tissue cells and viral infections, which is beneficial to resist viruses and reduce liver tissue lesions. Enhanced autophagy is conducive to MCMV viral replication and aggravates liver inflammation and the degree of pathological damage, whereas inhibited autophagy reduces viral replication and alleviates inflammation and liver pathological damage to some extent during this period of infection. Further understanding of the interactions between autophagy and MCMV infection and its potential mechanism may bring new and important clues to the control of MCMV infection or antiviral therapy.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (Nos. 81271807 and 81301425).