
Abstract
While an appropriately regulated production of interferons (IFNs) performs a fundamental role in the defense against coronaviruses such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), dysregulated overproduction of inflammatory mediators can play an important role in the development of SARS-CoV-2 infection-related complications, such as acute respiratory distress syndrome. As the principal constituents of innate immunity, both type I and III IFNs share antiviral features. However, important properties, including preferential expression at mucosal barriers (such as respiratory tract), local influences, lower receptor distribution, smaller target cell types, noninflammatory effects, and immunomodulatory impacts, were attributed only to type III IFNs. Accordingly, type III IFNs can establish an optimal effective antiviral response, without triggering exaggerated systemic inflammation that is generally attributed to the type I IFNs. However, some harmful effects were attributed to the III IFNs and there are also major differences between human and mouse concerning the immunomodulatory effects of III IFNs. Here, we describe the differential properties of type I and type III IFNs and present a model of IFN response during SARS-COV-2 infection, while highlighting the superior potential of type III IFNs in COVID-19.
Introduction
COVID-19
SARS-CoV-2, a member of the coronaviruses, is an enveloped virus with a positive sense single-stranded RNA as its genome (74,95). The principal structural proteins of SARS-CoV-2 are spike (S), membrane (M), envelope (E), nucleocapsid (N) proteins, and some accessory proteins (74). The S protein of SARS-CoV-2 plays a fundamental role in the viral entry into host cells. The SARS-CoV-2-related S protein binds to a human cell receptor, angiotensin-converting enzyme 2 (ACE2), with an affinity stronger (10–20 fold) compared with SARS-CoV (41). Thus, like SARS-CoV, the SARS-CoV-2 requires ACE2 to enter human cells (41,74). The ACE2 molecule is expressed in a low subgroup of lung cells, namely, type 2 alveolar cells (95). SARS-CoV-2 may also utilize other receptors or other cellular entry mechanisms, such as antibody-dependent internalization (95).
The COVID-19 clinical manifestations—fever, nonproductive cough, dyspnea, myalgia, fatigue, normal or reduced lymphocyte counts, and radiographic reports of pneumonia that resemble the symptoms of SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) infections (102,107)—emerge following an incubation stage of ∼2–14 days. The duration from the start of COVID-19 symptoms to death varied from 6 to 41 days with a median of 14 days. This duration is affected by the age and immune status of patients (102).
Susceptible subjects for COVID-19 are the individuals with underlying diseases including diabetes, hypertension, malignancy, chronic respiratory diseases, and cardiovascular diseases (30). Milder clinical symptoms were reported in children, when innate immunity may be highly effective (30). During initial stages of the COVID-19, the peripheral blood leukocyte count is normal or slightly low (39,124). In severe forms of the disease, the lymphocyte counts are significantly reduced (124). A strong association between inflammatory factors such as interleukin (IL)-6 and disease severity has been reported (33). Nonsurvivors exhibited an elevated number of neutrophils, and higher blood levels of urea nitrogen, creatinine, and
Although the mechanism of COVID-19 pathogenesis remains unclear, its semblances with SARS-CoV and MERS-CoV may provide mechanistic insights into COVID-19 pathogenesis (74). For example, the innate immunity-related inflammatory response and the virus-induced immunopathological events can play destructive roles during COVID-19 pathogenesis (12,19). The coronavirus-associated severe pneumonia is usually related to rapid replication of the virus, extensive accumulation of inflammatory cells, and elevated production of inflammatory mediators leading to acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) (12, 19).
The proper, balanced, and timely production of interferons (IFNs) can perform a fundamental role in limiting of a viral infection. However, inadequate- and untimely production of IFNs leading to the progression of viral infection, while overproduction of IFNs may lead to irreversible immunopathologic-mediated tissue damage. Herein, we present a model concerning the protective and deleterious roles of IFNs during SARS-CoV-2 infection and emphasize on the differential properties of type I (T1)- and type III (T3) IFNs to provide a novel insight into the therapeutic potentials of T3 IFNs in COVID-19 patients.
Type I and III IFNs
IFNs, as the first antiviral frontier, are categorized into three types, according to their receptor structure. T1 IFNs (IFN-α and IFN-β are main members besides IFN-δ, IFN-κ, IFN-ɛ, IFN-ζ, and IFN-ω) bind to an IFN-alpha receptor (IFNAR) complex, which consists of IFNAR1 and IFNAR2 chains, and T2 IFN (IFN-γ), the predominant immunoregulatory cytokine, signals through interferon-gamma receptor (IFNGR) (36,65,101,123). The members of T3 IFNs (named as IFN-λs) are IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4 bind to a receptor complex made up of IL-28RA and IL-10R2 (64,94,123).
Among the different types of IFNs, T1 IFNs can provide primary antiviral protection, since they are secreted by almost any type of cells after infection and can induce virtually any type of cells to confer protection (83). In contrast, IFN-γ does not exhibit this universal pattern of expression as its expression is limited to natural killer (NK) cells, T cells, and macrophages (4,51). Among the T3 IFNs, the IFN-λ3 exhibits the most powerful activity, followed by IFN-λ1 and IFN-λ2 (8,25,69).
The IFN-L4 gene is placed upstream of the IFN-L3 gene in the long arm of chromosome 19 (67). The amino acid similarity between IFN-λ4 and IFN-λ3 is about 30% (67). IFN-λ4 protein is expressed only by subjects carrying the IFNL4-ΔG genotype at rs368234815 (27). The in vitro experiments have indicated that IFN-λ4 is able to induce interferon-stimulated gene (ISGs) in HepG2 cells to the similar extent as IFN-λ3, and inhibits hepatitis C virus (HCV) replication (43,67). However, the IFNL4-ΔG has been associated with impaired clearance of HCV in virus-infected individuals, and several theories were presented to explain this controversy (27,67).
Production of T1 and T3 IFNs
The gene expression and the generation of T1 and T3 IFNs are triggered by similar stimuli (86). The pattern recognition receptors (PRRs) contributed to the induction of T3 IFN expression overlapping with those eliciting IFN-α and IFN-β expression. However, Ku70 (a cytosolic DNA sensor) selectively induces the IFN-λ expression, but not IFN-α and IFN-β expression (134). The cells of innate immunity, express PRRs that recognize the virus-originated pathogen-associated molecular patterns. For RNA viruses such as coronavirus, viral-related RNAs are identified by either the endosomal TLR3 and TLR7, or the cytosolic viral RNA sensors, such as melanoma differentiation-associated protein 5 (MDA5) and retinoic acid-inducible gene I (RIG-I). This recognition may eventually lead to the activation of transcription factors, including IRF-3, IRF-7, and nuclear factor-κB (NF-κB), which induce the expression of T1 and T3 IFNs, and other proinflammatory cytokines (123).
IFN-λ expression has a greater dependence on NF-κB than does IFN-α and IFN-β expression (46), indicating the differential characteristics of the gene promoter between T1 and T3 IFNs. Furthermore, T3 IFN expression is supported when the activated mitochondrial antiviral signaling protein (MAVS) is placed on the peroxisome, while IFN-β expression is promoted when the MAVS is placed on the mitochondria (69,89). The relative plenty of peroxisomes in epithelial cells can be a reason for the greater generation of T3 IFNs compared with IFN-β, in response to a viral infection (69). In hepatocytes, hepatitis B virus (HBV) infection triggers the T3 IFN expression (but not IFN-α and IFN-β) through a RIG-I- and MAVS-dependent manner (108), representing that the hepatocyte-specific elements may support the T3 IFN generation in response to some hepatotropic viruses.
T3 IFNs are secreted by some cell types, including, epithelial cells, macrophages, cytotoxic CD8+ T cells, NK cells, regulatory T cells, plasmacytoid dendritic cells (pDCs), conventional DCs (cDCs) and hepatocytes (86). T3 IFNs are the most abundant IFNs produced at mucosal barriers, such as respiratory-, gastrointestinal-, and urogenital tracts, following viral infections (86).
T1 and T3 IFN-mediated signaling pathways
Despite the use of different receptor complexes by T1 and T3 IFNs, the intracellular signaling pathways are substantially shared by these IFNs (120). Thus, T3 IFNs share antiviral activities with T1 IFNs. The signal transduction of T1 IFNs is initiated after the interaction between IFN and IFNAR, which triggers the activation of the relevant receptor-associated tyrosine kinases (TYK2 and JAK1). The activated JAK1 and TYK2 kinases phosphorylate the signal transducers and activators of transcription (STAT) molecules (123). The phosphorylation of STAT molecules triggers their homo- and heterodimerization, and their transportation into the nucleus. STAT1 and STAT2 are recognized as the principle IFN-stimulated transcription factors (120,123). The STAT1–STAT2 heterodimer binds to IRF-9 to form the ISG factor 3, which initiates the transcription of ISGs through binding to the IFN-stimulated response element in the ISG promoters. Many ISG-related products directly exert antiviral activities (123).
In humans, IFN-mediated transcription of ISGs leads to the expression of more than 7,000 genes, that are involved in some cellular processes, such as defense against viral infections, activation, migration, metabolism, and survival. Remarkably, many ISGs directly interfere with viral replication and transmission (123).
Although, T1 and T3 IFNs induce common signaling pathways, differential downstream signaling elements have also been explained. For example, JAK2 phosphorylation and activation are induced only by T3 IFNs but not by T1 IFNs (89,120). In spite of similar signaling pathways mediated by T1 and T3 IFNs, the earlier can induce ISG expression with a higher kinetic (earlier peak and faster decline) compared with T3 IFNs, which exert longer and more stable ISG expression than T1 IFNs (82,136). Thus, the T3 IFN-related impacts cause a delayed peak and a longer duration of ISG expression.
Target cells of T1 and T3 IFNs
The target cells for T1 and T3 IFNs differ. One of the major differences between the T1 and T3 IFNs is their receptor distribution. The T1 IFN receptor is commonly expressed in virtually all types of nucleated cells, but T3 IFN receptor expression is limited to very low cell types, such as epithelial cells of the respiratory, gastrointestinal, and urogenital systems, keratinocytes, hepatocytes, endothelial cells, and some subsets of innate immune cells (such as neutrophils, macrophages, cDCs, and pDCs) (4,120,136).
Among human immune cells, B cells exhibit much higher IFN-λR1 expression than that expressed by CD4+ and CD8+ T cells, whereas neutrophils, monocytes, and NK cells rarely express IFN-LR1 (106). CD8+ T cells exhibit greater expression of IFN-LR1 than CD4+ T cells. IFN-λ3 directly binds and upregulates ISG expression in human B lymphocytes and CD8+ T cells, but not in neutrophils, monocytes, NK cells, and CD4+ T cells (106). However, T cell receptor stimulation powerfully upregulates the expression of IFN-LR1 in CD4+ T cells. Thus, in activated CD4+ T cells, IFN-λ can induce ISGs and inhibit human immunodeficiency virus (HIV)-1 infection (106).
According to aforementioned explanations, T3 IFNs can directly interact with the human adaptive immune cells. In contrast to humans, the mouse neutrophils exhibit the highest expression of IFN-LR1 and other subsets of immune cells rarely express IFN-LR1 (10,32). Goel et al. also found that mouse splenic neutrophils respond to IFN-λ and upregulate ISGs, whereas human peripheral blood neutrophils fail to respond to IFN-λ (37). Human peripheral blood B cells express high IFN-λR levels and upregulate ISGs after stimulation with IFN-λ, whereas mouse splenic B cells fail to respond to IFN-λ (37). However, it has been observed when human neutrophils were stimulated with R848 (a synthetic TLR7 agonist) they can express IFN-LR1 (106). Thus, during an infection or inflammatory state, various types of immune cells can respond to T3 IFNs.
IFN-λR1 is constitutively expressed by epithelial cells of the respiratory system, and its expression is significantly raised immediately after viral infection (15). The preferential expression of the T3 IFN receptor on the mucosal barrier illustrates that T3 IFNs cooperate with T1 IFNs to act locally on epithelial and leukocytes to confer optimal immunity on epithelial surfaces with minimum tissue injury (10,32).
T3 IFNs also play an essential role in gastrointestinal-related mucosal immunity. In a mouse model of rotavirus infection, it was reported that T1 IFNs have no significant impacts on low IFNAR expressing gut epithelial cells (69,92). Intestinal epithelial cells powerfully respond to T3 IFNs to induce an antiviral response, as these cells express high amounts of IFNλR (69,92). T3 IFNs have weak effects on lamina propria cells that express low levels of IFNλR (80,92). However, Stanifer et al. showed that both IFN-β and IFN-λ can induce an effective antiviral state in SARS-CoV-2-infected human intestinal epithelial cell lines (115). Therefore, the study models (mouse or human), the virus types, the cell types, and the study conditions (in vitro or in vivo) should be considered for the results related to T1 and T3 IFNs.
The effects of T1 and T3 IFNs on leukocytes differ markedly, resulting in differential qualitative and quantitative extents of inflammatory reactions (121). T3 IFNs operate locally to trigger similar antiviral ISGs, while T1 IFNs mainly exert systemic influences (80,87). T3 IFNs primarily affect mucosal epithelial cells and protect them against viral invasion, without the deleterious inflammatory consequences attributed to T1 IFNs (121).
Immunomodulatory effects of T1 and T3 IFNs
In addition to inducing expression of antiviral ISGs, the T1 IFNs perform various effects on leukocytes, such as regulation of the differentiation, activation, and migration (123). IFN-α and IFN-β promote the expression of chemokines, CCL2, CCL3, CCL4, CCL5, CCL7, CCL12, CXCL9, CXCL10, CXCL11, whereas downregulate the expression of CXCL1 and CXCL2. The chemokines CCL2, CCL7, and CCL12 attract monocytes; CCL5, CXCL9, CXCL10, and CXCL11 induce T cell migration; CCL2 attracts memory T cells; CCL3 and CCL4 induce the migration of monocytes and macrophages; and CXCL1 and CXCL2 recruit neutrophils (53,123). Although, T1 IFNs inhibit the neutrophil recruitment by preventing CXCL1 and CXCL2 expression, IFN-α enhances the neutrophil survival by inducing the expression of cellular inhibitor of apoptosis.
T1 IFNs increase the NK cell activity, and increase the nitric oxide synthase expression in macrophages (118,123). T1 IFN-linked signaling in cDCs supports Th1 cell-mediated immunity (34). IFN-α and IFN-β enhance the expression of major histocompatibility complex molecules and the costimulatory molecules CD80, CD86, and CD40 (35,112). IFN-α and IFN-β increase perforin, granzyme B, and IFN-γ production in CD8+ cytotoxic T cells (18,123).
Because the T3 IFN receptor is expressed on restricted subsets of leukocytes, they may exert selective immunomodulatory activities. T3 IFNs perform pleiotropic effects on the immune system, a number of them highly resemble the T1 IFNs. T3 IFNs enhance the capacity of human monocyte-derived DCs to generate FOXP3+ regulatory T (Treg) cells (62,85). The results from in vivo experiments indicate that T3 IFNs suppress the capability of mouse DC2 cells to induce Th2- and Th17 cell differentiation and enhance the Th1 cell development (62). IFN-LR1 −/− mice exhibit reduced Th1 cell differentiation, whereas augmented Th2- and Th17 cell-related responses (62,120). Stimulation of peripheral blood mononuclear cells (PBMCs) in the presence of IFN-λ reduces the production of Th2 cell cytokines (including IL-4, IL-5, and IL-13), whereas increases the generation of IFN-γ indicating that T3 IFNs reinforce the Th1 cell-mediated responses (21,62).
T3 IFNs also trigger the generation of T1 IFNs and tumor necrosis factor alpha (TNF-α) and promote the expression of CD80 and CD86 in pDCs (29,84,120). Moreover, T3 IFNs directly inhibit mouse neutrophil-mediated proinflammatory activities by repressing the production of reactive oxygen species (ROS), downregulating of the neutrophil extracellular traposis (NETosis), inhibiting of the degranulation, and reducing the migratory capacity; these events limit the neutrophil-mediated tissue damages, and aid in maintaining the integrity of barriers (10,133). The impacts of T3 IFNs on mouse neutrophils are in sharp contrast to the proinflammatory effects of T1 IFNs, which activate neutrophils and other leukocytes (4,32). Although mouse neutrophils respond to both types of IFNs to potentiate the antiviral immunity, they exhibit proinflammatory properties only in response to T1 IFNs (4,32,123,138). Collectively, T3 IFNs seem to exert immunoregulatory activities, as they possess the antiviral features of T1 IFNs, whereas they lack some of the proinflammatory properties of T1 IFNs. Thus, T3 IFNs perform a principle role in immunoregulation and provide sufficient protection with minimum tissue damage (86,120) (Table 1). An emerging function of T3 IFNs is the regulation of innate immunity. Unlike T1 IFNs, the T3 IFNs suppress proinflammatory responses and limit the host-damaging effects associated with inflammation (32). These properties are largely attributed to their selective inhibitory impacts on mouse neutrophil-related proinflammatory functions (10).
Comparison of the Type I Interferon- and Type III Interferon-Related Properties
IFN, interferon; IFNAR, IFN-alpha receptor; IL, interleukin; ROS, reactive oxygen species; NK, natural killer; MAVS, mitochondrial antiviral signaling protein; ISG, interferon-stimulated gene; DC, dendritic cell; pDC, plasmacytoid DC; cDC, conventional DC; MHC, major histocompatibility complex; NOS, nitric oxide synthase; TNF-α, tumor necrosis factor α; Treg, regulatory T; NETosis, neutrophil extracellular traposis.
Antiviral effects of T1 and T3 IFNs
T3 IFNs exhibit antiviral activity against a range of viruses, such as HBV, HCV, herpes simplex virus, yellow fever virus, HIV, Zika virus, vaccinia virus, influenza A virus, influenza B virus, SARS-CoV, human metapneumovirus, and respiratory syncytial virus (RSV) (69). However, the in vivo antiviral impacts of T3 IFNs are more effective against viruses that infect the epithelial cells of the respiratory-, gastrointestinal-, and urogenital system, as well as the liver (3,69).
The in vitro analyses showed that the primary epithelial cells of the nose, bronchi, and alveoli express high concentrations of T3 IFNs, but not T1 IFNs, after infection with respiratory viruses, such as influenza A virus, measles virus, RSV, and mumps virus (4,90,125). The bronchial smooth muscle cells also express T3 IFNs in response to infection with rhinovirus (5). T3 IFNs inhibit rhinovirus replication in bronchial epithelial cells (69). The production of T3 IFNs is inversely associated with the viral burden and disease severity in persons experimentally infected with the rhinovirus (69).
In a mouse model of influenza A virus infection, the magnitude expression of T3 IFNs is negatively associated with the viral load and the disease severity (56). Early produced T3 IFNs are essential to prevent influenza A virus transmission from the upper airways to the lungs, a process that takes place early during infection (32,61). As respiratory system-related epithelial cells, certain types of residential and infiltrated leukocytes into the lung express the receptor for T3 IFNs, these IFNs can act on the respiratory epithelial cells and local leukocytes to limit the initial propagation of the influenza A virus infection (32).
According to animal models of viral infections, T3 IFNs can forgive initial local antiviral defense without triggering unnecessary systemic inflammation. It has been speculated that T1 IFNs may act as the second border of defense, after viruses escaping from T3 IFNs' control (32). T1 IFNs may systemically limit viral infection, whereas T3 IFNs can locally control infection at mucosal and epithelial barriers (80,87). Therefore, T3 IFNs may be regarded as regulators of the antiviral system that have been developed to prevent unnecessarily excessive T1 IFN-linked aggravated inflammation (86,128). For example, during influenza A virus infection, T3 IFNs prevent the generation of TNF-α, IL-1β, and other tissue destructive mediators in mouse neutrophils, while enabling the development of efficient antiviral responses (32).
The therapeutic potentials of T3 IFNs have been highlighted in animal models with influenza A virus infection and colitis (24,32). T3 IFNs provide a protective role in an animal model of colitis through a fine suppressive impact on the production of ROS by neutrophils, protecting the intestinal mucosal during acute inflammation (10). Moreover, the therapeutic potentials of T3 IFNs have been indicated in other diseases such as collagen-induced arthritis (7) and arterial thrombosis (13). In an animal model of arthritis, the efficiency of the T3 IFNs therapy was attributed to their inhibitory impacts on Th17 cells and neutrophil migration (7).
According to the results from studies using animal models of infection with respiratory viruses (especially influenza A virus) it has been concluded that in the respiratory system, both T1 and T3 IFNs are generated by epithelial cells that can maintain an antiviral function through the autocrine and paracrine impacts (17,121). If a dominant T3 IFN response occurs during a respiratory viral infection, an efficient antiviral response is established that control the viral infection without systemic inflammatory responses (121). Because T1 IFNs exert pleiotropic effects, numerous cell types are activated that cause a massive unnecessary inflammatory response with serious consequences (121).
The IFN-λR knockout mice exhibit increased susceptibility to respiratory viruses, including influenza virus, RSV, and SARS-CoV virus (69). Influenza virus infection in IFN-λR knockout mice causes the greater viral load, elevated neutrophil, and total leukocyte counts in the bronchoalveolar lavage fluid (BALF), and worsen lung efficiency due to tissue damage (32). T3 IFNs neutralization exacerbates acute influenza virus infection in mice with normal IFN-β expression, while intranasal administration of T3 IFNs effectively suppresses infection with influenza A virus and related consequences (24,59).
In contrast to T3 IFNs, the treatment of influenza A virus-infected mice with IFN-α increases the lung proinflammatory cytokine levels, induces neutrophil recruitment, and causes epithelial cell death (24,59). The in vitro experiments indicated that human and mouse airway epithelial cells were responders to IFN-α and T3 IFN by expression of antiviral genes without expression of the proinflammatory cytokines, whereas PBMCs responded only to IFN-α (24,59).
IFN Response During Coronavirus Infections
After entering its host, the SARS-CoV-2 may pass through the nasal and laryngeal mucous membranes, reaches the lungs through the respiratory tract. After that, the virus can enter the blood from the infected lungs, leading to viremia. SARS-CoV-2 then attacks the ACE2-expressing organs such as heart, kidney, and gastrointestinal tract (72). Massive SARS-CoV-2 replication, enormous death of the epithelial and endothelial cells through apoptosis, vascular leaks, diminished ACE2 expression, malfunction of the renin–angiotensin system, hyperinflammatory responses, and cytokine storm play fundamental roles in the COVID-19 pathogenesis (31).
In SARS and MERS mouse models, the delayed response of type I IFNs disrupts the viral control followed by hyperinflammation, extensive lung infiltration by neutrophils and macrophages, apoptosis of epithelial and endothelial cells, which ultimately leads to ARDS (11,68). Thus, an initial IFN response exerts protective influences in SARS-CoV- and MERS-CoV-infected mice and the delayed response of T1 IFNs disrupt the immunity against SARS-CoV and MERS-CoV (11).
In a mouse model of SARS-CoV infection, irregular T1 IFN response and activation of the inflammatory monocytes–macrophages are the main causes of lethal pneumonia (12). It was proposed that after SARS-CoV infection, delayed T1 IFN would disrupt the early control of viral infection, causing accumulation of inflammatory neutrophils and monocytes–macrophages that is manifested in lung immunopathology, including ARDS and pneumonia (11,12). In animal models of SARS-CoV and MERS-CoV infections, when T1 IFNs have been used for treatment, the timing of administration is a key parameter that influences the outcome response (12,95). According to the results from a mouse model of SARS-CoV, it has been concluded the targeting of T1 IFNs using relevant receptor blockers or antagonists should be considered as an option to ameliorate the harmful inflammatory responses during later stages of SARS disease (11).
Heterogeneous patterns of T1 IFN's response were reported in COVID-19 patients as some studies indicated impaired response, while others found raised IFN response. The in vitro and animal model experiments also revealed that SARS-CoV-2 triggers a weak T1 IFN response in comparison with other pulmonary RNA viruses (6,14). SARS-CoV-2 did not substantially trigger T1, T2, or T3 IFN response in the human pulmonary tissues (14).
The mild and moderate types of the SARS-CoV-2 infection have been associated with a powerful type I IFN response characterized by the potent ISG expression in the BALF of COVID-19 patients (55). Furthermore, patients with mild/moderate forms of COVID-19 exhibit a greater type I IFN response during 8–12 days compared with severe patients (55). Accordingly, the clinical outcome of COVID-19 may be influenced by the time and extent of IFN response. Trouillet-Assant et al. reported impaired T1 IFN response in some critically COVID-19 patients, which was associated with a poor outcome (119). Hadjadj et al. reported undetectable plasma IFN-β levels and low IFN-α levels in severe and critical patients, which were correlated with blood viral load and aggravated inflammatory reactions (42). The blood number of pDCs as the main producers of T1 IFN was reduced in severe patients with COVID-19 as compared with mild/moderate patients (42). However, whole blood cells respond normally to IFN-α indicating that the responsiveness to T1 IFN was intact in COVID-19 patients (42).
On the other side, it was revealed that the monocytes from patients with severe COVID-19 exhibited higher expression T1 IFNs accompanied with higher expression of proinflammatory cytokines TNF-α/IL-1β compared with milder COVID-19 (70). Thus, T1 IFNs may reinforce hyperinflammatory response in COVID-19 through promoting TNF-α/IL-1β expression (70). Zhou et al. also indicated that SARS-CoV-2 infection powerfully induced the expression of a number of ISGs accompanied by the expression of proinflammatory cytokines and chemokines in BALF samples collected from COVID-19 patients (137). Collectively, it seems that an initial appropriate T1 IFN response can limit viral expansion, whereas delayed T1 IFN response may promote pathological responses.
Based on the experience obtained from SARS-CoV and MERS-CoV infections, the IFN-α and IFN-β have been considered as potentially effective therapeutic agents against SARS-CoV-2 (55,105). It has been emphasized that T1 IFN should be implemented as soon as possible following infection (ideally before initiation of symptoms) but not at late stages to prevent tissue damage (38,55). The results from a meta-analysis study indicate that treatment of COVID-19 patients with T1 IFN reduces the mortality rates and improves discharge times (122). Several types of T1 IFNs (such as NCT04320238, NCT04293887, and ChiCTR2000029989) are being evaluated for clinical efficacy (55).
The kinetic of the T3 IFNs during COVID-19 remains to be explained in future studies. As mentioned above, during the infection with influenza A virus, T3 IFNs are the earlier IFNs that are produced in the respiratory mucosa, confer viral resistance, and limit the initial viral spreading (32). In a mouse model of influenza A virus infection, it was observed when the viral load is low, the T3 IFN response is enough to limit infection. However, when the viral load is high in the first stage or the virus evades the T3 IFN defense, then the T1 IFNs are triggered to complete antiviral protection (4,32). Upon influenza A virus infection of the airway epithelial cells, T3 IFNs are secreted first to operate as the first border of defense to control the virus spreading at the epithelial barrier without stimulating systemic inflammation (32). If the virus escapes the T3 IFN line defense, then the T1 IFNs are produced that provide the second line of protection, enhance the antiviral immunity beyond the respiratory epithelium and stimulate the proinflammatory cascades that are essential for protection but also leading to immunopathologic reactions (4,32).
In this study, we propose a model concerning the T3 IFN response during COVID-19, which is based on the experience obtained from other respiratory viral infections. Clinically, the IFN responses during infection with SARS-CoV-2 can be classified into two phases (Fig. 1). During the incubation time of COVID-19, an efficient IFN response may be required to eliminate the virus and to prevent disease progression to moderate and severe stages.
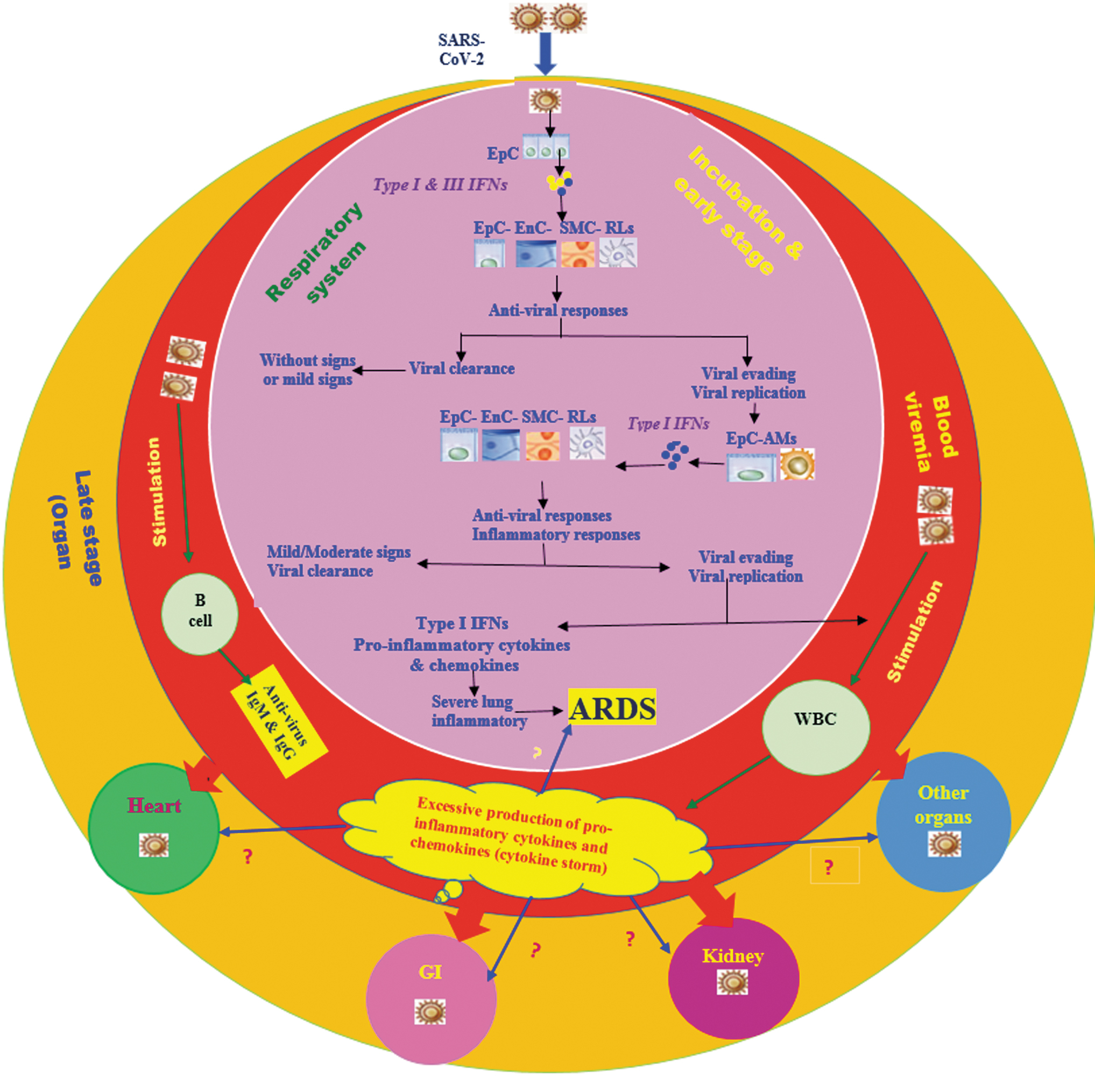
A proposed model concerning the stages of SARS-CoV-2 infection and the possible IFN response. During incubation and early stages of COVID-19 the respiratory system may be primarily affected. After inhalation, SARS-CoV-2 infects the nasal-, larynx-, and lung epithelial cells. The SARS-CoV-2-infected epithelial cells may produce T1 and T3 IFNs (mainly T3 IFNs) that can provide an initial antiviral response through influencing various types of residential cells. This initial antiviral response may cause viral clearance without expression of clinical symptoms or may be accompanied by mild respiratory symptoms. In high proportions of the SARS-CoV-2-infected subjects, the virus is eliminated at this stage. However, in some SARS-CoV-2-infected subjects, the virus may evade the initial antiviral IFN response and replicate. Presumably, this primary immune evading can lead to second antiviral IFN-(greater production of T1 IFNs) and inflammatory responses. The second antiviral IFN- and inflammatory responses may cause viral clearance accompanied with mild/moderate symptoms. In some SARS-CoV-2-infected subjects, the virus may be eliminated at this stage. However, in a low number of infected subjects SARS-CoV-2 can evade the second antiviral IFN response and spread, enter the blood from the lungs, and cause blood viremia. During the blood stages, SARS-CoV-2 can activate various types of WBCs causing cytokine storm, which is a dangerous systemic inflammatory response arising from the production of large quantities of proinflammatory mediators. Cytokine storm exerts serious destructive effects on several systems, for example, in the lungs causing ARDS development. SARS-CoV-2 may enter multiple organs from the blood and cause organ failure during the late stage of COVID-19. IFN, interferon; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; ARDS, acute respiratory distress syndrome; EpC, epithelial cells; EnC, endothelial cells; SMC, smooth muscle cells; RLs, residential leukocytes; AMs, alveolar macrophages; WBC, white blood cells.
In the incubation phase, if an appropriate local IFN response (first T3 IFN and then T1 IFN) is induced in the upper parts of the respiratory system, SARS-CoV-2 may then be efficiently eliminated without or with mild clinical symptoms. Therefore, the strategies for boosting the immune responses at the early stage are certainly important. Thus, in the COVID-19 patients without symptoms or with mild symptoms, an effective local T3 IFN response is induced that eliminates the viral infection without pathogenesis. Indeed, more than 80.0% of the SARS-CoV-2-infected patients display mild signs (30), however, a low proportion of COVID-19 patients exhibit severe clinical symptoms (44). When the initial IFN response fails, the virus spreads, enters the blood, and causes massive destruction of the infected tissues, especially in organs that express high amounts of ACE2, such as kidney and intestine. The presence of SARS-CoV-2 was indicated in autopsy or biopsy specimens obtained from several organs such as heart, liver, kidney, and intestinal epithelium (76,113,116,126). Inadequate local IFN response in the respiratory system may lead to virus persistence and replication. If the patient is immunosuppressed or immunodeficient, combined with underlying diseases, the immune system cannot effectively control the virus in the incubation phase. The virus enters the blood (viremia phase) from the lungs and migrates to the intestine and kidney. In this situation, the hyper-expression of the T1 IFNs and other pro-inflammatory cytokines and chemokines can lead to the occurrence of the cytokine storm phenomenon that plays a major role in the development of the SARS-CoV-2-related complications (Fig. 1).
The T1 IFNs reinforce inflammatory responses characterized by the upregulation of various kinds of cytokines and chemokines, such as TNF-α, IL-1β, and IL-6, whereas T3 IFNs lack this impact (4). T3 IFNs induce only the expression of ISGs without affecting the production of inflammatory mediators (4,32). Accordingly, the recombinant T3 IFN administration in experimental animal models of influenza A virus suppresses the inflammatory cascade triggered by the respiratory viral infection, whereas IFN-α exerts the opposite effect (24,32). In mouse, the neutrophils are the key point of the antiviral and/or proinflammatory activities of T1 and T3 IFNs. The mouse neutrophils respond to both T1 and T3 IFNs to potentiate the antiviral protection, however, they exhibit proinflammatory properties, solely in response to T1 IFNs (32,123,138).
Therefore, overexpression of T1 IFNs with high infiltration of inflammatory monocytes/macrophages and neutrophils may play a crucial role in lung dysfunction during the late stage of COVID-19 (102,110). Once severe lung damage occurs, attempts should be taken to reduce inflammation and to manage the symptoms (110).
Coronaviruses Evade IFN Response
The appropriate T1 IFN response can be protective in SARS-CoV and MERS-CoV infections, however, in both infections the T1 IFN-mediated antiviral responses are suppressed (60,111).
SARS-CoV and MERS-CoV employ various strategies to inactivate signals leading to the T1 IFN production and/or the signals that cause ISG expression. The damping of the T1 IFN-mediated antiviral responses is strongly related to the disease severity (12,95). SARS-CoV and MERS-CoV can trigger the formation of the vesicles that lack PRRs, thus preventing the recognition of their dsRNA by host PRRs (114). During the induction phase of T1 IFNs, SARS-CoV and MERS-CoV interfere with the signaling pathways located downstream of cytoplasmic RNA sensors (111). SARS-CoV and MERS-CoV have been equipped with mechanisms to prevent the STAT-1, IRF-3, and IRF-7 activation (60). Both structural (such as M, N) and nonstructural open reading frame (ORF) proteins of SARS-CoV and MERS-CoV contribute to the suppression of T1 IFN response (60,95).
Accessory protein 4a of MERS-CoV interferes with IFN expression by preventing the MDA5 activation (through direct interaction with double-stranded RNA) and blocks the IFN-mediated ISG expression by suppressing the protein kinase R-related pathways (88,98,111). Moreover, ORF4a, ORF4b, ORF5, and M proteins of MERS-CoV prevent IRF3 and IRF7 activation, and block IFN-β production (60,131).
A variety of mechanisms are used by DNA and RNA viruses to block the T3 IFN-mediated responses. For example, the E3L protein of vaccinia virus, the VP24 protein of Ebola viruses, a viral protease of coxsackievirus, the NS1 protein of norovirus, the Vpr and Vif proteins of HIV-1, interfere with the expression and T3 IFN-mediated signaling pathways (136).
The results from in vitro experiments have indicated that SARS-COV-2 is more vulnerable to T1 IFNs than SARS-COV (55,77). SARS-CoV-2 does not interfere with T1 IFN-mediated activation of STAT-1 (55,77). The remarkably lower T1 IFN response and Th1 cell activity have been reported in a COVID-19 patient with eventual death than in a recovered patient (28). SARS-CoV-2 may utilize similar strategies to modulate the T1 and T3 IFN responses (95). For example, SARS-CoV-2-related ORF3b acts as a powerful inhibitor of T1 IFN production (63). SARS-CoV-2-derived proteins, such as Nsp1, Nsp2, Nsp3, Nsp12, Nsp13, Nsp15, PLpro, ORF3a, ORF3b, ORF6, and ORF9b interfere with antiviral T1 IFN response (48,71,103). The targeting of the IFN response escape pathways used by SARS-CoV-2 is essential for effective management of the COVID-19 (74).
The Cytokine Storm in COVID-19
Cytokine storm, a destructive inflammatory response, arising from the excessive secretion of proinflammatory cytokines and chemokines, is accompanied with the exacerbation of COVID-19 exhibitions (74,95). Elevated serum quantities of IFN-γ, TNF-α, IL-1β, IL-2, IL-7, IL-8, IL-9, IL-10, IL-17, granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), IP-10, MCP1, MIP1A, and MIP1B were measured in COVID-19 patients (45). Severe COVID-19 patients exhibit higher quantities of TNF-α, IL-2, IL-7, IL-10, G-CSF, CXCL10, MCP1, and MIP1A in comparison with the patients display milder forms (45). The cytokine storm is considered as the main etiological mechanism for ARDS development (74,95).
Cytokine storm can facilitate viral sepsis and promote inflammation-mediated lung injury, which cause serious complications, such as pneumonitis, ARDS, respiratory abnormalities, shock, multiple organ failure, and eventually death, in severe cases of SARS-CoV-2 infection, just like those that occur in SARS-CoV and MERS-CoV infections (74,95). Lung epithelial and endothelial cell apoptosis is one of the earliest outcomes of the cytokine storm (12). IFN-α, IFN-β, and IFN-γ induce the infiltration of inflammatory cells and cause apoptosis in the airway and alveolar epithelial cells (12). The inflammatory macrophage-derived TNF-α also promotes apoptosis in the lung epithelial and endothelial cells causing vascular leakage and alveolar edema, eventually leading to hypoxia (12).
Severe COVID-19 patients suffer from lymphopenia and main cytokine storm players, such as TNF-α and IL-6, can contribute to lymphopenia through induction of apoptosis in lymphocytes and through direct inhibitory effects on hematopoietic progenitor cells, respectively (12,54,78,79). As coronavirus-specific T cell-mediated responses play a key role in viral elimination, the lymphopenia helps viral persistence (54,135).
Many cytokines that are involved in the cytokine storm are associated with the Th17 cell-mediated responses (47,52). IL-17 influences the neutrophil differentiation (by stimulating G-CSF and GM-CSF secretion), recruits neutrophils (by inducing neutrophil-attractant chemokines, such as CXCL1, CXCL2, and CXCL8), recruits other inflammatory cells (by inducing IP-10, MIP2A, and MIP3A), and triggers tissue damage and remodeling (by inducing matrix metalloproteinases) (129). IL-1β, IL-6, and TNF-α cause systemic inflammatory symptoms, including fever and C-reactive protein production, which are clinical signs of COVID-19 (45,95). IL-22, another Th17 cell-derived cytokine, may contribute to the formation of life-threatening edema that is mixed with mucus and fibrin, seen in SARS-CoV-2 and SARS-CoV patients (129).
High numbers of CCR6+ Th17 cells were reported in the peripheral blood sample from a patient with severe COVID-19 (130), further supporting the involvement of Th17 cell-related cytokines in the cytokine storm development. Elevated responses of Th17 cells have also been observed in MERS-CoV and SARS-CoV patients (28,58). In MERS-CoV patients, higher amounts of IL-17 with lower amounts of IFN-γ and IFN-α have been associated with worse outcome (28). Elevated levels of Th17- and Th1 cell-related cytokines were also detected in serious infection with the H1N1 influenza virus (58). Infection of mice with a certain strain of the H1N1 influenza virus causes ALI in an IL-17-dependent manner (73). Collectively, the Th17 cell-linked responses may contribute to the cytokine storm development in the SARS-CoV-2 infection, which may cause tissue damage and promote pulmonary edema. Therefore, targeting of Th17 cell-related pathways may have beneficial effects in severe forms of COVID-19 to attenuate the harmful cytokine storm. Indeed, the preventive effects of T3 IFNs on the Th17 cell activity were reported, which may have attenuating impacts on the cytokine storm (62,120). According to the results from mouse models of influenza A virus infection, T3 IFNs have the potentials to prevent cytokine storm (4).
Some Major Considerations About T3 IFNs
We emphasize that both the beneficial and detrimental effects of T3 IFN signaling are mainly obtained from the results of mouse studies. Therefore, some results from mouse studies may not be generalizable to humans, as there are main differences between mouse and human regarding the T3 IFN-related immunomodulatory effects. For example, the beneficial impacts of T3 IFNs in mouse respiratory viral infections largely attributed to their selective inhibitory effects on neutrophil-related proinflammatory activities (10). T3 IFNs directly affect the mouse neutrophil proinflammatory function, by suppressing the ROS production and degranulation of neutrophils, thereby they limit the neutrophil-mediated tissue damage and help the maintenance of the barrier integrity (10).
Unlike mice, human neutrophils rarely express IFN-λR1 and do not respond to T3 IFN (37,106). However, LPS and R848-treated human neutrophils can express IFN-λR1 (106). If during a human respiratory viral infection, neutrophils express IFN-λR1, they can respond to T3 IFNs. Moreover, human peripheral B cells express IFN-λR1, whereas mouse splenic B cells fail to respond to IFN-λ (37).
Second, in influenza A virus-infected mice, it was found that treatment with T1 and T3 IFNs during the recovery period (7–10 days after infection) disrupts lung repair (81). IFN-β- and IFN-λ-induced P53 directly decreases the expansion and differentiation of airway epithelial cells (9,81). Investigations using IFN-LR1−/− mice also indicated that endogenous IFN-λ prevents epithelium regeneration during influenza recovery, through direct deleterious effects on epithelial cells and increased susceptibility to bacterial superinfection (81). Using a mouse model, it was also observed that prolonged IFN-λ production by lung DCs in response to a synthetic viral RNA causes epithelium damage and inhibits tissue repair by direct impacts on epithelial cell expansion and viability (9).
Third, prolonged IFN-λ exposure also promotes susceptibility to bacterial superinfections in the inflamed lungs (9). According to mouse experiments, it was concluded that T1 and T3 IFN-related signaling can promote respiratory superinfection mainly through influencing on inflammasome, Th17 cells, neutrophils, and macrophages, as well as the nasal microbiota colonization (91). Treatment with T3 IFN declines neutrophils in BALF and enhances bacterial load during influenza superinfection in a mouse model (100).
Although, early IFN-λ administration in a mouse model of SARS-CoV-2 infection can confer protection (26), the results from mouse studies should be carefully evaluated in humans. The duration and timing of IFN-λ administration should also be carefully chosen for any therapeutic attempts in COVID-19 treatment.
Conclusion
Since coronaviruses are capable to evade the host immune responses, thus their control and elimination require therapeutic interventions such as using IFNs. Both T1 and T3 IFNs induce a similar class of antiviral genes, but T1 IFN exhibit powerful proinflammatory features, as a wide range of cell types are responsive to stimulation by T1 IFNs (Table 1). Thus, they may also exacerbate viral-associated complications by promoting excessive inflammatory responses.
In SARS-CoV-infected mice, it has been indicated that the early T1 IFN response is protective, whereas delayed T1 IFN signaling impairs antiviral immunity (11,12). Hence, using T1 IFN antagonists has been recommended as an intervention to control unnecessary detrimental inflammatory reactions during later phases of severe SARS-CoV infection (11). The timing of IFN therapy is a very important parameter influencing the disease outcome. At later stages of SARS-CoV-2 infection, the possible promoting effects of T1 IFNs on the cytokine storm, as the main etiological mechanism for COVID-19-related complications (such as ARDS) may be a serious concern.
T3 IFNs confer local antiviral immunity at mucosal barriers, without inducing potent systemic proinflammatory properties attributed to T1 IFNs (7,24). Coronaviruses such as SARS-CoV-2 mainly infect the respiratory epithelial cells and T3 IFNs may stimulate antiviral genes in the lung epithelial cells, without inducing excessive inflammatory responses.
The unique biological properties of the T3 IFNs mentioned above make them as attractive therapeutic agents in COVID-19 patients. Pegylated IFN-λ1 is already accessible and is now in Phase 3 evaluations for hepatitis delta virus. The results from in vitro experiments have indicated that the IFN-λ inhibits the SARS-CoV-1 and MERS-CoV replication (93). In vitro evaluation also indicated that IFN-λ1 can strongly prevent the replication of SARS-CoV-2 in human airway epithelial cells (26). In a mouse model of SARS-CoV-2 infection, the therapeutic and prophylactic administration of pegylated IFN-λ1 decreases viral replication (26). Therefore, the clinical use of IFN-λ in COVID-19 may be very promising, and clinical trials using Pegylated IFN-λ1 (NCT04343976, NCT04354259, and NCT04331899) have been started (93).
T3 IFNs may be used in combination with other antiviral drugs and supportive medications (129). Experimentally, the beneficial effects of T3 IFNs in a number of inflammatory and allergic disorders have also been reported (86,120). The possible treatment with T3 IFNs need standardization concerning the doses, route of administration, timing, stages of viral infection, and underlying diseases.
It is considerable that adaptive immunity components, including CD8+ cytotoxic T lymphocytes, effector CD4+ T cells (Th1-, Th2-, Th9-, Th17-, and Treg cells), and B cells may play differential roles during the different stages of SARS-CoV-2 infection. Therefore, the impacts of the exogenous T3 IFNs on the adaptive immune responses to SARS-CoV-2 need to be cleared in future investigations.
Moreover, the association of the several genetic polymorphisms in the T3 IFN locus with viral clearance and favorable outcomes has been indicated in some viral infections such as HBV, dengue virus, HCV, and HSV-1(69). The presence of CC genotype in IFN-λ3 rs12979860 single nucleotide polymorphism (SNP), and the presence of TT CC genotype in IFN-λ3 rs8099917 SNP were associated with protection against hepatocellular carcinoma development among patients with HCV and HBV infection (97). The presence of the T allele in IFN-λ3 rs12979860 SNP was also related to severe symptoms in dengue virus-infected children (20). Moreover, an association was indicated between the presence of the TT genotype at IFN-λ3 rs8103142 SNP and IFN-λ2 rs12980602 SNP with protection against HCV infection, viral load, and outcome in HCV-infected subjects (132). Furthermore, the CC genotype of IFN-λ3 rs12979860 SNP and the TT genotype of IFN-λ3 rs8099917 SNP were more prevalent in patients with chronic HCV infection (96).
SNPs can influence the gene expression, mRNA stability, the half-life of cytokine, and its affinity for receptor binding (49,50). Some SNPs may be in preferential linkage with other functional SNPs. For example, the IFN-λ3 rs4803217 SNP influence mRNA stability. The rs12979860, rs12979860, and rs8099917 were associated with diminished expression of IFN-λ3 during HCV infection. The impact of the rs368234815 SNP on the IFN-λ4 expression is well documented (27). The possible association of the gene polymorphisms in T3 IFN locus with disease severity, expression of the clinical symptoms, response to a given therapeutic program, and clinical outcome of COVID-19 needs to be examined in further investigations.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.