
Abstract
Despite advances in slowing the progression of acquired immunodeficiency syndrome (AIDS), there is no viable cure for human immunodeficiency virus (HIV). The challenge toward a cure is mainly the formation and maintenance of a latent reservoir of cells that harbor the virus in both replication-competent and replication-defective states. This small niche of quiescent cells has been identified to reside primarily in quiescent and memory CD4+ T cells, but parameters that could reliably distinguish an infected T cell from an uninfected one, if any, are not clear. In addition, the migratory properties and specific anatomical reservoirs of latent T cells are difficult to measure at a high resolution in humans. A functional cure of HIV would require targeting this population using innovative new clinical strategies. One constraint toward the empirical development of such approaches is the absence of a native small animal model for AIDS. Since HIV does not efficiently infect murine cells, probing molecular-genetic questions involving latently infected T cells homing to deep tissue sites, interacting with stroma and persisting through different treatment regimens, is challenging. The goal of this article is to discuss how examining the dynamics of T cells in mouse models can provide a framework for effectively studying these questions, even without infecting mice with HIV. The inflammatory and cytokine milieu found in early human HIV infections are being increasingly understood as a result of clinical measurements. Mouse studies that recreate this milieu can potentially be used to subsequently map the fate of T cells activated in this context as well as their migratory routes. In essence, such a framework could allow complementary studies in mice to enhance our understanding of aspects of the biology of HIV latency. This can be the basis of a modular approach to small animal HIV modeling, amenable to preclinical curative strategy development.
Introduction
The progression of human immunodeficiency virus (HIV) infections to acquired immunodeficiency syndrome (AIDS) has been thwarted quite effectively in the clinic by current regimens of antiretroviral therapy (ART). Particularly, the use of combined ART (cART), rather than monotherapy, has improved the lifespan of HIV-infected patients from ∼12 years to 55 years for patients diagnosed at age 20 (47). The next looming challenge is the complete cure of HIV, requiring elimination of all copies of the reactivatable virus, which persists anywhere in the body, even in cART-treated patients. This is even more pressing in the developing world, where multiple factors, including cost, logistics, and social dynamics, limit ART administration. A seemingly impenetrable obstacle to a cure is HIV latency, or the presence of a reservoir of quiescent cells, which harbor viral genomes, often in a defective state, even when actively replicating viruses are eliminated by ART cocktails (30, 63, 66, 93).
The challenges posed by this reservoir require a concerted basic science approach aimed at understanding the exact mechanisms behind their formation, maintenance, and therefore potential avenues for their control and functional elimination. Human and Non-Human Primate (NHP) studies have made remarkable progress in recent years, improving our understanding of the generation of latent HIV reservoirs in patients and their relationship to the dynamics of T cell responses in vivo. While these are encouraging, the lack of a small animal model has been a significant limitation (16, 84). Mouse models, as in other fields, including tumor immunotherapy, typically allow for high-resolution cellular, molecular-genetic, and pharmacological manipulations that are not easy to do even in NHP models. The power this affords for dissecting basic biological principles using gain and loss of function studies, as well as in high-throughput development of therapeutics, is a welcome complement to HIV cure research. In this article, we discuss how reductionist approaches using existing mouse models are relevant for preclinical development of latency-targeting strategies even without an infectious virus model in mice.
The core idea is that given the wealth of clinical measurements available from HIV infections in humans (and NHP studies), it is now possible to recreate specific aspects of the presentation as “modules” in mouse strains. Combining multiple such reductionist modules offers an alternate strategy to reconstruct the process of latency, even without an actual HIV infection, and use it for cure research. It should be pointed out that in the clinical setting of human infections, there is an emerging consensus that a functional cure, but not complete elimination of the virus, is a more realistic goal. Nevertheless, this need not be the bar in the reductionistic models. Ideally, approaches developed in preclinical models that offer the highest degree of infected cell elimination would be most relevant to porting to clinical strategies for a functional cure.
While some of the findings that make it possible to consider such modular approaches are in fact quite old, others are more recent. It has been known for a while that HIV primarily infects activated CD4+ T cells and that the long-term reservoir of latent viruses is also thought to largely persist in the memory T cells that follow (24). These memory-phenotype cells have often been characterized as “quiescent,” a reflection of a lowered metabolic and transcriptional state of the T cells. These are not actively proliferating cells, but still capable of migrating in response to chemokines. This state is not induced by viral infection, rather it is the result of a coordinated contraction of the activation-generated effector T cells into a pool of memory T cells (66, 93). Of course, the generation and maintenance of a quiescent pool of memory T cells are part of the normal biology of an immune response, even without a latent HIV infection (57). Certainly, understanding the dynamics of these natural memory populations can help us also understand how latently infected T cells behave and expose Achilles heels that can be exploited for curative strategies when such cells are also HIV reservoirs.
Consistent with heterogeneities in HIV tropism during the early infection phases, other cell types and subsets can also contribute to the reservoir (54, 89).These include (but are not limited to) monocyte-derived macrophages (17), embryonically derived macrophages (59), follicular dendritic cells (DCs) in lymphoid tissue (99), microglia (71), and other types still being determined. Macrophages, in particular, have been reported to be not only a potential source of viral persistence but also contributors to the re-emergence of competent virus after ART discontinuation (42, 52, 114).
Even within the T cell compartment, CD4+ T follicular helper cells have emerged as a subtype that harbors latent virus and may in fact contribute to the insidiously ongoing persistent viral replication in infected individuals with undetectable viral levels (18, 70, 73). This may be due to a protective environment in germinal centers and follicles, where these cells reside, which prevent effective killing of virally infected cells (41). While non-T cell types may indeed contribute to the total viral load in infected people, recent studies highlight how the T cell reservoir presents a unique and major concern (93).
There are also reports of T cells with a naive phenotype (107, 108) or those that may have been activated, but still resemble naive T cells (25) carrying latent virus. While there is discussion in the field about these data, the important point related to this article is in their migratory properties. Naive T cells typically sample the secondary lymphoid organs for antigen and rarely transit through or much less stay in other tissues (112). In contrast, as part of the normal biology of memory T cells, they are initially activated, proliferate in the secondary lymphoid organs, but then can migrate throughout the entire body to occupy multiple deep-tissue niches, and survive for a human lifetime (57, 102). During this time, these cells can also proliferate multiple times, inadvertently expanding the latent viral genomes they carry (53).
Accordingly, while a successful curative strategy for HIV relies on eliminating the virus, it is really in understanding the several properties of these different T cells that host them, that mouse models can offer potential insight, which may be hard to glean from observational human and NHP analyses. While we do discuss some aspects of HIV latency in this context, more comprehensive survey of the latency literature itself can be found in several recent reviews (63, 66, 93, 105).
HIV Infection, T Cell Activation, and Latency: A Complex Dance
The lifecycle of HIV, including the generation of latent infections, is intimately coupled with the physiological dynamics of peripheral CD4 T cell activation. T cells found in the peripheral lymphoid organs after completing development in the thymus, but have never undergone activation by their target antigen, are therefore naive T cells. In children and young adults, naive T cells constitute the bulk of the T lymphocyte population. It is generally agreed that such naive cells are not hospitable for HIV infection—but this is not a universal view (25, 107, 108). Instead, productive HIV infection is typically thought to require the host T cell to first undergo activation. Typically, T cell activation is a tightly regulated process, requiring a series of signals from the antigen-presenting cell (APC)—starting with a target antigen presented on Major Histocompatibility Complex (MHC) molecules.
In addition, the overall milieu around the APC during early activation of T cells is quite critical in fully activating the T cell and guiding its differentiation trajectory all the way to memory T cells. The various combinations of molecular signals present in this milieu can also program distinct long-term fates in the activated T cells (57). Such fates not only include the differentiation toward various effector cytokine productions (typically referred to as Th1, Th2, etc.) but also include the migratory properties and survival in the effector/memory phases (55, 68, 98). At a molecular level, these changes involve the T cells changing the expression of various genes, including transcription factors, chemokine receptors, and adhesion molecules, in response to instruction from the activation milieu.
Once fully differentiated, these effector T cells can migrate to every tissue in the body, ostensibly to fight localized infections or malignancies. Importantly, these migratory routes are governed by the contextual signals that T cells receive during the early activation phase. For instance, signals from gut-derived DCs through retinoid acid drive up the expression of chemokine receptors and integrins that promote homing to the gut, while different cues from lung-derived DCs during early T cell activation promote homing to the lung (38, 87).
The consensus view is that CD4 T cells are infected during activation (and proliferation through the effector phase) of the naive T cell and this has been extensively reviewed (74). Optimal functional infection and integration of the viral genome into host cell DNA require host cell activation. It should be noted that viral entry into resting T cells has been demonstrated under in vitro conditions (26, 28, 115). This has been recently characterized as “preactivation” latency (direct establishment in resting memory cells) and “postactivation” latency; Rezaei et al. have shown that these two modes of latency establishment result in quantitatively differing amounts of reactivatable virus (85). Current latency modeling and detection tools that rely on fluorescent reporter systems may underestimate the preactivated subset contributing to the reservoir (61).
Although earlier studies focused on the latent reservoir being seeded early after HIV infection (9, 29), recent work using phylogenetic modeling from pre- and post-ART human donor samples does indicate additional and later entry into the latent pool (3, 22). These studies emphasize the need to parallel modular mouse modeling with human studies. For instance, a separate model, developed to mimic the late milieu could be valuable in assessing if the whole body fate of cells seeded later in the course of HIV are different from the early ones.
The nature of the virus that integrates into the host genome has also been better understood. These typically fall into two broad categories—intact versus defective proviruses. When viewing the latent reservoir from the perspective of potential for viremic emergence, cells that contain replication competent virus are the most concerning (111). In studies analyzing the total latent and replication-competent pool of HIV, such as the Swiss cohort study, recurrent blips of viral replication are noted (12). These events tended to be quite rare and did not significantly modify the slope of viral decay over a 5-year period.
However, from human and NHP studies tracking the dynamics of these virus forms, it appears that (at least under conditions of ART) the behavior of cells carrying either intact or defective forms is quite similar (12, 23, 110). An extended discussion of this phenomenon and its applicability to a “rinse and replace” strategy for cure was recently published (46). A creative extension of these arguments is that it should be possible to now model the fate of a latently infected T cell, even in an animal model where viral replication is not possible. This module would only focus on the host cell, but with proper mirroring of the data obtained from human and NHP studies, it could be quite valuable, as discussed below.
To mirror the contextual cues around the T cell that is getting infected by HIV (and potentially committing to a long-term latently infected reservoir fate as well), pinpointing the original infection of the latent cell is important. Since the initial activating milieu would imprint an “area code,” comprising of a specific combination of chemokine receptors and cell adhesion molecules on the activated T cell, which allows it to preferentially home to the appropriate tissue site, the expectation is that modeling the “HIV milieu” in a T cell activated in a mouse model can inform us about the potential subsequent trajectory of the activated cell. This could allow the porting of existing knowledge about T cell behavior in vivo directly to the study and manipulation of HIV reservoir dynamics.
Learning More About the Fate of Latent HIV Reservoir, from Basic Mouse Models
Indeed, expanding on the above logic then, for a defined set of parameters associated with early HIV infection, it might even be possible to predict the long-term fate of potential latency host T cells using our current knowledge of T cell activation dynamics. This fate prediction would include the migratory patterns of the HIV host cells, their metabolic states, long-term survival, and so on, all of which would be of great help in developing new generations of therapies targeting these cells and aimed at eradicating latently infected cells. Obviously, the foundational high-resolution modeling required to map the establishment of latency in humans to this degree is problematic both technically and logistically.
Unfortunately, no small animal model currently exists that perfectly recapitulates HIV pathogenesis in a human environment. NHPs can provide, and have provided, invaluable advancements in developing HIV therapeutics and understanding pathogenesis; particularly, in macaques, recent research has attempted to explore topics such as viral seeding, latency establishment, and the effects of particular cellular subsets in viral control (64, 76). However, NHP models do not feasibly approach the manipulative power of small animal models, such as the mouse model (Box 1). In a modular approach, availability of NHP models is a useful intermediary.
Box 1: Sample of existing mouse models:
Mice are useful as relatively inexpensive small animal models of complex physio/pathophysiological conditions. A primary challenge with HIV and mice lies in the inability for HIV to naturally infect and progress in wild-type mice as it does in primates (1, 2). While the focus of this article is to discuss how the process of HIV latency may be modeled in mice without a HIV infection, we summarize previous themes used to model HIV infections in mice:
It is important to point out that approaches with mouse models and NHP occupy different spaces of research. We are not extensively discussing the latter since their utility is quite well documented. Indeed, NHP studies would be critical for preliminary hypothesis derived from modular mouse models to be tested rigorously. Toward this end, infections of SIV or the combined simian-human immunodeficiency virus (SHIV), which can be used in NHP models, can be valuable (15, 64). For the purposes of this review, we will focus our discussion on the use of mouse models, which have the distinct ability to be manipulated on a shorter timescale, and in greater numbers. The key question is how such models can be leveraged, given that, as we have discussed, HIV does not infect any genetically tractable laboratory host.
We have so far highlighted some challenges in finding the right animal model for exploring HIV pathogenesis and the establishment of the latent reservoir. A solution might lie in combining reductionist approaches, focusing on two parallel processes that take advantage of current understanding of T cell biology, as opposed to trying to optimize specific HIV infection models for non-human species—(a) the precise measurement of the cellular milieu during the initial HIV infection and (b) the inference in small animal models of how such milieu can program T cell fate. While progress in both fronts is being made, it is likely that combining the available information will also allow us to identify the pieces that need to be filled in, toward such a predictive methodology.
The initial milieu, in which early HIV dissemination and infection occur, is around the mucosa. We know quite a bit already about the cells in this milieu and the changing environment as the HIV infection progresses, mostly from studies in NHP models. Brenchley et al. demonstrated that there is a preferential depletion of CCR5+ (a marker of activation) CD4+ T cells at mucosal sites in the gastrointestinal tract during HIV infection, whereas CD4+ T cells in lymph nodes proliferate and account for the replenishing of susceptible T cells for infection (20).
Mucosal perturbation during early HIV infection was further shown by observing higher levels of local proinflammatory markers, such as microbially derived lipopolysaccharides, in untreated individuals (21). Several studies have found an elevation of plasma IL-6 during acute HIV infection, stemming from activated monocytes; in the context of HIV, this is associated with B cell activation, as well as an increase in all-cause mortality (19, 77). Concurrently, there is extensive literature exploring how IL-6 impacts CD4+ T cell fate: IL-6 can positively impact T cell survival, differentiation into Th17 subset (and transition away from a Th1 subset), and secretion of IL-21, among other downstream effects (35). C-reactive protein, which is similar to IL-6 in its significance as an acute-phase protein secreted during acute infection, has also been shown to sway CD4 T cells away from a Th1 differentiation (116).
Another interesting molecule that appears to be elevated during initial HIV infection is Galectin-9, which is a soluble lectin that some studies have shown to activate T cells through T cell receptor (TCR) signaling modulation, as well as increase the secretion of proinflammatory molecules (such as IL-2 and TNF-α) (1, 33). TNF-α is an important hallmark of acute infection, which can be secreted primarily by monocytes and macrophages; in vitro studies have demonstrated that this increase in TNF-α results in a macrophage-specific decrease in CCR5 surface expression, which, as we have discussed, is a crucial receptor of HIV entry into T cells (67, 80).
Finally, plasmacytoid DCs in the mucosa have been demonstrated to sense HIV RNA through innate receptors, and subsequently secrete type I interferons; this may increase CD4 T cell migration to sites of infection, as well as impact the differentiation state of those recruited T cells (78). The above observations have been summarized in Figure 1. It should, however, be emphasized that many of the above data are derived from in vitro experiments or NHP models; to test the production of inflammatory molecules in acute HIV infection in humans is challenging considering the lack of symptomology during that time of disease.

Inflammatory context of early HIV infection drives immunological fate. During the context of HIV infection, a milieu of cytokines and inflammatory molecules act to shape the environment. These factors drive changes in T cell migration, differentiation, and overall fate. While not a conclusive list, this figure is meant to highlight some of the observed molecules that drive this environmental shift and illustrate the complexities of the HIV-driven inflammatory environment. We propose that understanding the range of factors that shape this environment can allow for researchers to more accurately glean how the pool of susceptible T cells toward latent infection might emerge early on in the course of the virus. HIV, human immunodeficiency virus.
In addition to molecules that are released as a product of an innate response to infectious perturbation or immune cell activation, there are also damage-associated molecular patterns that activate pattern recognition receptors on innate immune cells to induce an inflammatory response (86). Indeed, there is some evidence that cellular damage associated with initial HIV infection, including the release of ATP from dying cells, can act on purinergic receptors found on CD4 T cells to prime them for HIV entry (94).
The next part of a modular strategy for developing models for HIV is to use some of this information and mirror that in a mouse model. This could range from simple treatment experiments where activated T cells are exposed to the local environment mimicking the HIV infection context in vivo to transgenic experiments where these milieu and triggers are built in genetically.
Along these lines, there are several elements from the milieu that can be already studied in mouse models. For instance, it is known that the tropism of HIV generates a curious scenario, in which the very cells that respond to active infection express the co-receptor (CCR5) for HIV entry (79). Sites that are known to have higher amounts of CCR5+ (recently activated) T cells are therefore theoretically more susceptible to HIV infection; mucosal effector sites fit this characterization quite nicely; however, other tissue sites are known to attract these CCR5+ T cells (65, 79).
Indeed, inducible expression of such receptors on mouse T cells can be used to mimic these biases as well. Since latency involves defective viruses in CD4 T cells, we argue that the actual viral replication of HIV in small animal models may not be as critical a component. For instance, even using a self-inactivating lentivirus infection in the context of a reconstituted inflammatory milieu mirroring known HIV-induced components, we can manipulate the variables necessary for tracking the fate of potential host cells that would normally harbor latent HIV. This reductionist approach has been used in other infectious models that are difficult to port into small animal models; for example, in the current SARS-CoV-2 pandemic, attenuated viral strains & mouse-adapted versions are currently being explored as potential animal models to examine vaccine design, pathogenesis, and other viral traits difficult to glean from the nonattenuated existing viral models (69).
However, the impact of ongoing viral replication on the latent pool may also be possible to approach by modeling as a separate module. As emerging data from analysis of gene expression in the human HIV reservoir (11, 32, 101, 113) are further parsed out, we can now include those molecular details. Conditional overexpression of IRFs or inducible apoptotic genes could, for instance, recapitulate some downstream consequences of viral replication. Nevertheless, the absence of viral replication in these models will need to be considered in translational stages, with NHP and human studies.
While this is an incomplete list of contextual information relevant to the early milieu in which HIV latency gets established, it already raises several questions amenable to a reductionist enquiry in mouse models. For instance, how does mucosal inflammation associated with microbial translocation, or high IL-6, affect T cell fate, specifically in the context of retroviral infection? It should be relatively straightforward to recapitulate such an environment, even without an HIV infection in the animal model—and track marked T cells.
A key question then would be to be able to map these findings back to NHP models and human infections. There are several notable techniques that have emerged recently to track antigen-specific T cells spatially through their migration to sites of inflammation; for example, 64Cu uptake of T cells by the TCR coupled with positron-emission-tomography tracking has been demonstrated to work in vivo (4, 45). Several studies have very recently used fluorescent tracking of HIV-infected cells as a high-sensitivity in vivo method to track latency in small animal models (109). Many of these can be applied to NHP models with SIV or SHIV to validate findings from modular approaches.
The Uniqueness (or Lack Thereof) of the Latent Reservoir
Understanding the trafficking patterns and niches of latently infected T cells is perhaps only a first step toward a cure—the key is to remove these cells from all the niches they occupy (46). This, of course, would require us to (a) understand how to target the survival mechanism of the latently infected cells and (b) devise strategies to interfere with these in clinical settings. Currently, neither is well defined. However, again, an important clue—which is amenable to reductionist modeling—is that HIV seems to hide in T cells that are naturally long lived in secondary lymphoid organs and perhaps in deep tissue sites (92).
After the clearance of an acute infection, memory T cells have evolved multiple mechanisms that allow them to remain in the body in a relatively quiescent state for a very long time (39, 81). Data supporting the persistence of memory T cells have converged from multiple studies. In humans, naive and memory T cells can be distinguished by quantitating the TCR rearrangement excision circle (TREC). During the genetic events leading to the development of the TCR, a circular DNA piece is excised from the genome (14, 49). This remains stably in the cell as TREC, but since it is only generated once in the life of a cell, it is diluted (or lost to some of the progeny) when the T cell divides. Thus, TREC dilution within a population of T cells is a measure of activation and proliferation in human T cells (36). Indeed, Douek et al. used a PCR-based assay to show that memory T cells lacked detectable amplification of TRECs, compared to naive T cells—due to TREC dilution (36).
There is also evidence to suggest that the bias toward longevity in memory T cells is accompanied by certain metabolic characteristics of these cells, compared to an effector/activated population; memory T cells exhibit the use of oxidative phosphorylation and fatty acid oxidation, whereas activated effector cells generally employ a glycolytic pathway (10, 43, 62). Glycolytic pathways have been illustrated to negatively impact T cell longevity and persistence through interaction of intracellular signaling survival pathways (62).
Despite the observed small percentage of quiescently infected memory cells, a study from Siliciano et al. found that these cells have an extremely long half-life, with a mean of 44 months in patients undergoing HAART (97). This would mean that from time of infection, and assuming consistent HAART usage, it would take an estimated 73.4 years to lose 106 latently infected CD4 memory T cells (97).
That is not to diminish the usage of HAART. In fact, pre-exposure prophylaxis (PrEP), which involves the use of HAART in at-risk HIV-negative individuals, has been shown to provide a significant reduction in HIV seropositivity when joined with strategies that increase HAART adherence and affordability (37, 40, 100). Adherence to PrEP remains a consistent obstacle in raising the overall efficacy of this prevention measure. While an extensive discussion of PrEP is beyond the scope of this perspective, a reader interested in understanding HIV-Cure strategies is well served by reading other reviews (31, 48, 88, 100). ART administration is quite prohibitive.
Given these data, it is important to note that a significant fraction of our current understanding of memory T cells in humans is derived from mouse studies. So the model systems required to study them are already in place. Layering contextual information from HIV studies to such models is relatively straightforward. The change in perspective as related to HIV cure is that while most mouse studies are currently focused on understanding how memory generation and maintenance can be enhanced, the perspective for HIV cure is the opposite. There are, as yet, no good methods to eliminate some of all of these cells in vivo, although one was discussed recently from a theoretical vantage (46).
Memory T cells are a very heterogeneous population both in terms of cell phenotype and inhabited niches. It is prudent to point out that efforts to identify unique markers identifying infected cells in humans are ongoing; there is, to date, no consensus of unique features that distinguish them. In 2017, Descours et al. had found evidence that the low-affinity Ig receptor FCγRIIa (CD32a) was upregulated significantly in quiescently infected CD4 T cells (34). However, attempts to recapitulate these findings have been met with little to no success; sorting for CD32a does not reliably illustrate an enrichment of latently infected CD4+ T cells (82). Furthermore, other studies have identified upregulation of CD32a on CD4+ T cells; however, this appears to reflect generalized T cell activation rather than a unique property related to the HIV reservoir (13). While we discuss a general strategy to port aspects of HIV to mouse models, these markers, once validated, could make it much easier to target them in humans directly.
Developing New Paradigms for Tracking and Targeting the Latent Reservoir
Considering these challenges, reductionist approaches offer a different vantage. As we have noted, there is a contextual environment of initial acute inflammation that drives T cell migration and differentiation into quiescent cells. A power of mouse models can be used to examine the spectrum of diversity that follows from activated cells that were in an equivalent milieu (Fig. 2). Indeed, both human and mouse studies offer clues to developing such models for HIV latency relevant studies (90).
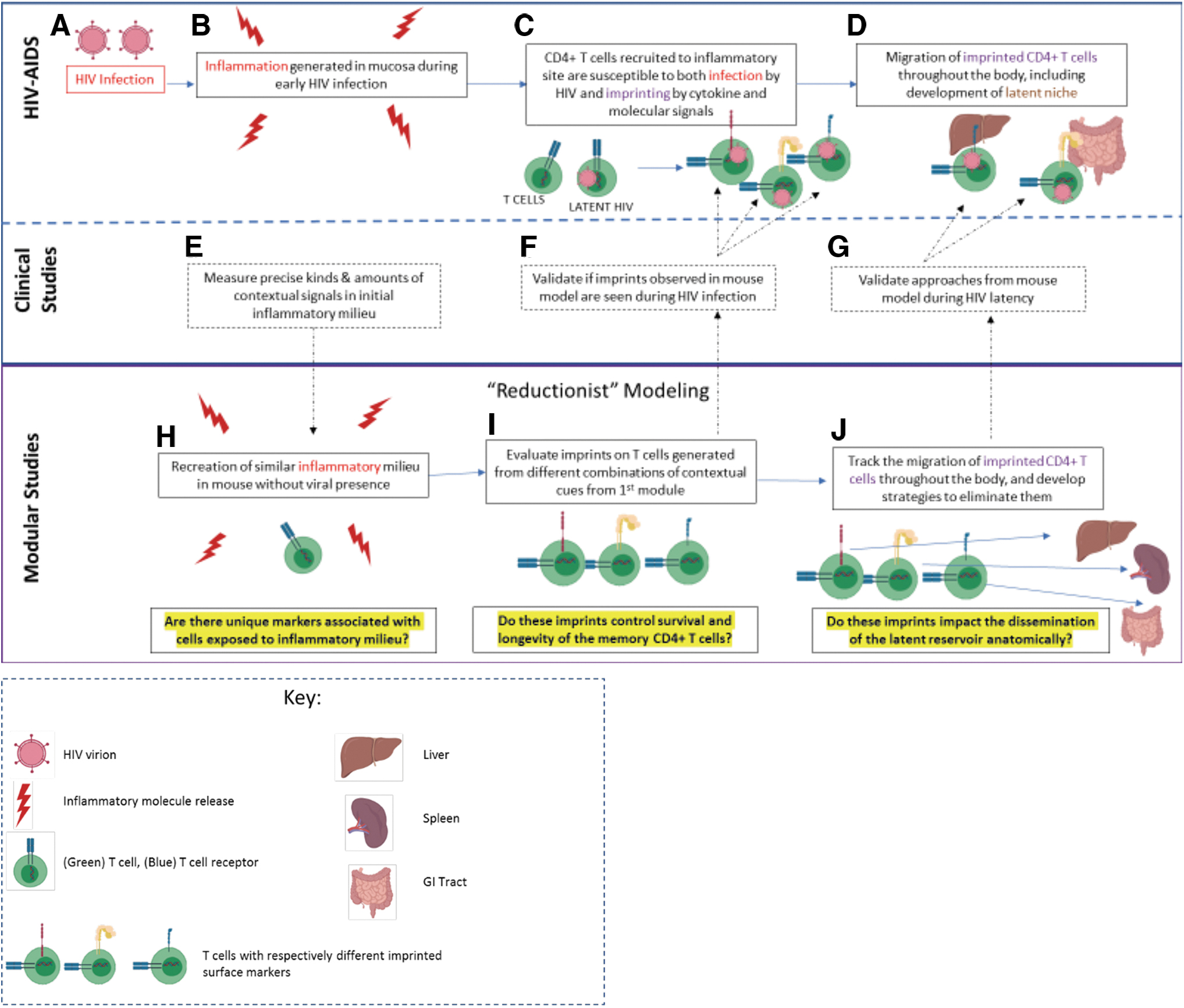
Parallel approaches toward modeling the cellular dynamics of HIV latency relating clinical data to animal models.
Technology exists using transgenic TCRs, fluorescent markers, barcoded DNA, and/or specific peptide; MHC conjugates would bind to specific TCRs within mouse models to specifically detect and track populations of responding T cells over time and physiological distance (44, 45, 104). Very recently, Neidleman et al. described a method of identifying latently infected cells using mass cytometry; importantly, they combined this technique with analytical tools to identify precursor latent cells without using reactivation, something that more closely resembles the homeostatic quiescent nature of the reservoir (75).
Moving forward, techniques such as these will be important for understanding the biology of the reservoir, but are limited in the challenge to collect large amounts of quiescent latently infected cells. In this sense, we propose to recapitulate the transition into “latently” identified cells without using an actual HIV infection; rather, if we recreate the environmental cues that occur during viral infection, we can model immunologically how some cells become suited for latency.
While these kinds of studies will allow us to develop a high-resolution description of potential fates that latent cells can adopt, the next challenge is to develop approaches to eliminate them. The need for new frameworks is especially acute now, given the challenges with the current paradigms (46). The most prominent of such efforts was termed “Shock and Kill,” a concept revolving around using chemical agents to reactivate a resting T cell (providing the “shock”), thereby allowing the newly activated T cells to be targeted by antiviral immune effects (93). This theory is derived from observations that T cell transcription factors (NFκB for example), which play a role in T cell activation, bind and enhance the transcription of HIV (50, 93).
While the shock and kill approach addresses the necessity for the elimination of the quiescent pool of infected T cells, the main difficulty lies in finding a latency reversal drug that is strong enough to reactivate the infected cells without inducing systemic toxicity. Furthermore, a hallmark study by Ho et al. found that not only do many viruses remain noninduced following stimulation but also a significant portion of these noninduced viruses are replication competent. This study additionally illustrated that the original size of the latent reservoir, determined through mitogenic stimulation to be a mere few cells out of 1 × 106 CD4 T cells in the lymph node or blood, may in fact be much larger (51, 56).
Other strategies have also recently emerged, which have targeted latency for eradication without the “shock” component of cellular reactivation. One such strategy has been termed “Lock and Block,” in which the main goal is to permanently induce latency among infected cells, thereby not allowing for any re-emergence into a viremic state (2, 106). While many of the proposed therapeutics for Lock and Block are still being explored in promising in vitro and in vivo studies, this strategy faces the big challenge of finding the right drug that can selectively target latently infected cells (5). Ultimately, as we have indicated, this fundamental question concerning the identity, maintenance, and persistence of the latent reservoir continues to haunt the field of HIV research. This highlights once more the need to understand the factors that go into shaping the reservoir, and as we have outlined, this may be accomplished by reviewing the context of the initial inflammatory infectious event.
Conclusions
We have discussed thus far the utility of using a small animal model to explore the features that govern a transition into a quiescently infected T cell. While we have focused the bulk of the discussion on activated “memory phenotype” T cells, it is important to note that data on naive and other T cell states that may be targeted by HIV can be gleaned from such approaches as well. However, as we have also indicated, modeling a human disease in non-human organisms can present challenges, but these discrepancies between the host-pathogen relationships are a common concern in most animal models. While such models are important in unraveling basic molecular pathways as well as developing new reagents to target such pathways, they are hardly precise replicas of the clinical condition. Even when using pathogens such as influenza virus or malarial parasite to initiate the pathology under study in an animal model, prudent curation of the findings is typically required before preclinical and translational application.
The context of HIV, at first blush, seems different because the virus does not infect cells in a small animal model. We discuss in this article how this is not necessarily a handicap; indeed, conscientious modeling of different modules of HIV infection even without a virus can be quite informative. Therefore, we have discussed a reductionist approach, one that aims to determine the physiological conditions that occur during acute HIV infection, which ultimately shape the transition to latency—the beauty of this approach is the ability to reconstruct the initial inflammatory milieu that shapes the fate of the T cell, without needing to use an organism-specific HIV construct. We also discuss briefly other cell subsets (in addition to memory CD4+ T cells) that have been cited as potential contributor to the reservoir.
Even though we chose to focus our model on memory CD4+ T cells, we believe that the concept of generating a reductionist approach based on the initial inflammatory context shaping the fate of the immune system can be applied broadly to understand other cell fates, including the naive T cells or the macrophage reservoir that has recently been elaborated. For our purposes, this approach allows us to manipulate individual variables of that inflammatory milieu individually and examine how each component contributes to the determination of the T cell's fate. To this end, we believe that the story surrounding latency and HIV is inextricably intertwined with that of T cell activation and migration, and that only with a proper understanding of the latter, can we make further progress into eliminating the virus entirely from a host.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
National Institute of Allergy and Infectious Diseases (R21 AI149076-01A1).