
Abstract
The nuclear factor-kappa B (NF-κB) signaling network constitutes a first line of defense against the invading viruses. However, viruses also adopted multiple strategies to interfere with NF-κB activation. Enterovirus 71 (EV71), in the family Picornaviridae, has become the main pathogen responsible for hand, foot, and mouth disease. Recent studies have reported that the nonstructural protein 2C of EV71 inhibits TNF-α induced NF-κB activation by suppressing IKKβ phosphorylation. In our study, we found that 2C can form inclusion bodies (IBs) in infected and transfected cells. Furthermore, 2C was able to sequester IKKβ into IBs through direct interaction with IKKβ. Although 2C did not directly interact with IKKα, viral protein 2C was able to sequester the IKKα into the IBs mediated by IKKβ. Our in vitro data further demonstrated that EV71 2C could suppress IKKα phosphorylation. These all together support a novel mechanism for EV71 to escape from NF-κB response, in which the phosphorylation of IKKα was suppressed by being recruited into viral IBs in the presence of 2C and IKKβ.
Introduction
Enterovirus 71 (EV71) contains a single-stranded positive-sense RNA genome, which is frequently associated with hand, foot, and mouth disease and most likely to occur in children of age 2–4 (2). The genome of EV71 is about 7.5 kilobases, which encodes four structural viral proteins (VP1, VP2, VP3, and VP4) and seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) (15). The 2C protein is one of the most conserved viral proteins among all picornaviruses. It contains an N-terminal membrane-binding motif, an adenosine triphosphatase (ATPase) domain, a cysteine-rich motif, and RNA-binding sites (5). The 2C protein participates in many critical events throughout the life cycle of EV71. It is not only essential for viral replication, but also plays an important role in evading host antiviral responses by interfering with nuclear factor-kappa B (NF-κB) pathway (4). Zheng et al., have shown that EV71 2C suppresses IKKβ phosphorylation through interaction with IKKβ and blocks NF-κB responses (22). IKKβ together with IKKα and IKKγ are the three main kinase subunits of the IκB kinase (IKK) complex of NF-κB pathway (14). The phosphorylated IKKα and IKKβ would lead to the phosphorylation of the IκB proteins, which are inhibitors of NF-κB transcription factor (17).
Viral inclusion bodies (IBs) are a special structure in cells, formed through viral proteins integrating with host cell protein, organelle membranes, and intracellular lipid droplets (11). The shape, assembly components, and formation mechanism of viral IBs vary greatly (11). IBs are supposed to play a role in immune escape response (21). In our study, we observed that EV71 infection promoted IBs formation in the presence of 2C. IKKβ localized in IBs and colocalized with 2C through direct interacting with IKKβ. We further demonstrated that IKKα translocated into IBs mediated by IKKβ in the presence of 2C, which lead to the suppression of IKKα phosphorylation. Altogether, we proposed a novel host immune evasion strategy for EV71, which involves the recruitment of IKKβ and IKKα into IBs to suppress IKKα phosphorylation.
Materials and Methods
Approval was received for application for Laboratory Animal Welfare and Ethical review of Nanjing University (SYXK SU 2019-0056).
Cell lines and virus infection
HeLa cell lines (ATCC) and Human embryonic kidney 293T (293T) (ATCC) cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (GIBCO, Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (GIBCO). Cells were maintained in an environment of 37°C and 5% CO2. Cells were tested for mycoplasma contamination and authenticated through FACS analysis (GIBCO). For confocal microscopy assays, the cells were grown on glass coverslips. The EV71 virus strain Fuyang-0805 used in this study was kindly provided by Dr. Wu Bin (Jiangsu Provincial Center for Disease Control and Prevention). For infection, the viruses were inoculated in HeLa or 293T cells in DMEM free of FBS. After 2 h uptake at 37°C, the inoculum was replaced by fresh DMEM containing 2% FBS. The cells were collected at different time points for analysis.
Antibodies and cytokines
Anti-IKKα and anti-IKKβ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p-IKKα and anti-p-IKKβ were purchased from Abcam (Cambridge, MA). Mouse anti-β-actin antibodies were obtained from Cell Signaling Technology (Danvers, MA). Anti-Flag, anti-hemagglutinin (HA) and anti-EGFP were purchased from Santa Cruz Biotechnology. Secondary antibodies, used for western blot analyses and confocal microscopy, were supplied by Santa Cruz Biotechnology or Abcam, respectively, and 4, 6-diamidino-2-phenylindole (DAPI) was obtained from Sigma-Aldrich (St. Louis, MO). Human rTNFα was purchased from BioVision (San Francisco, CA). EV71 2C antibody (Catalog no. GTX132354) was obtained from GeneTex Company.
Construction of plasmids
2C cDNA was subcloned into plasmid pRK5 with HA tag for mammalian expression under a cytomegalovirus promoter. The primer sequences of 2C are as follows: 5′ primer (BamH I), 5′-CGGGATCCAAGCGCTTCCTGGCTCAAG, and 3′ primer (Sal I), 5′-ACGCGTCGACTTATTGGAAAAGAGCC. The primer sequences of 3D are as follows: 5′ primer (EcoR I), 5′-GGAATTCGGGGAGATCCAGTGGGTTAA; and 3′ primer (BamH I), 5′-CGGGATCCTTACTAAAATAACTCGAGC. HA-3C, Flag-IKKβ, Flag-IKKα, HA-IKKα, and EGFP-IKKβ were kindly provided by Thomas Leung (Institute of Molecular and Cell Biology, Singapore). HeLa cells or 293T cells were transfected with various plasmids with Lipofectamine 2000 reagents following the manufacturer's instructions.
Immunofluorescence microscopy
HeLa or 293T cells grown on glass slides were cotransfected with plasmids 2C, 3C, 3D, IKKβ, and IKKα. At indicated time points, slides were washed twice with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 30 min, and permeabilized with 0.1% Triton X-100 for 10 min. Cells were blocked with 5% bovine serum albumin (BSA) at 37°C for 1 h. Following washing with PBS twice, cells were incubated with primary antibodies (1:100) in PBS containing 1% BSA at 4°C overnight. After three washes with PBS, cells were subsequently incubated with appropriate secondary antibodies (1:200) at 37°C for 1 h and stained with DAPI (1 μg/mL). Slides were mounted with #1.5 cover slips and Mowiol mounting medium and examined using an FV3000 confocal microscope.
Transient transfection, immunoprecipitation, and western blotting assays
293T cells were transfected with HA-2C alone or with Flag-IKKβ, Flag-IKKα for 24 h. The cells were harvested and lysed with a precooled 1% NP-40 lysis buffer containing 10 mM HEPES (pH7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 2 mM PMSF, 2 mM NaF, 1 mM Na3VO4, 1 g/mL aprotinin, and 1 g/mL leupeptin on ice for 20 min. Cell lysates were obtained by centrifugation at 12,000 rpm and 4°C for 5 min. Then, cells were incubated with either specific or control antibodies at room temperature for 2 h and then precipitated with protein A/G beads. After incubation, beads were washed four times with lysis buffer. Protein concentration of the lysates was quantified using the BCA Protein Assay Kit (Pierce, Rockford, IL) before loading for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Sample proteins were separated using SDS-PAGE before protein immobilization on polyvinylidene difluoride membranes (Millipore). Membranes were blocked with 5% nonfat milk and probed with primary antibodies, followed by overnight incubation at 4°C. After washes, the membranes were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody. Images were captured using FluorChem FC2 Imaging System (Alpha Innotech Cooperation, San Leandro, CA) and developed following the manufacturer's instruction.
Statistical analysis
Data are expressed as means ± standard deviations from three independent experiments. The samples were performed in triplicate. One-way analysis of variance was used for comparison among the different groups by the SPSS software. For statistical analysis, a two-tailed Student's t-test was used to evaluate the data by SPSS software (IBM SPSS, Armonk, NY). p-Values <0.05 were considered statistically significant.
Results
EV71 infection forms viral IBs in the presence of 2C
The composition of virus-induced IBs differs substantially, even between closely related viruses. Therefore, there is no well-established marker for viral IBs so far. The well-recognized diagnosis protocol for viral IBs is based on visualization of foci formation. Here, we have used anti-2C antibody to visualize viral IB (red) in EV71-infected HeLa cells and 293T cells. HeLa cells were infected with EV71 strain Fuyang-0805 at an MOI of 1. The cells were collected at 6, 12, and 24 h postinfection and subjected to immunofluorescence microscopy using anti-EV71 2C antibody to visualize viral IB (red), respectively. Foci in the cytoplasm were observed in the EV71-infected cells, which represented the viral IB. Moreover, the size of IBs became bigger along with the EV71 infection time (Fig. 1A, C). However, the number of IBs increased at 6 and 12 h not at 24 h. At 24 h, we observed that some IBs were merged with each other to become bigger ones. IBs were also observed in EV71-infected 293T cells (Fig. 1D–F), which suggested that IBs formation upon EV71 infection applied to different cell types.

IBs formed by 2C in EV71-infected cells. HeLa
EV71 2C contributes to IBs formation
To understand the role of 2C in the formation of IBs, we examined its localization in 2C-overexpressed HeLa cells without EV71 infection. The 2C cDNA were cloned in an expression vector and transfected into the HeLa cells for 24 h. As shown in Figure 2A, viral protein 2C first appeared as granules and later changed into patches, which corresponded to the IBs as observed in Figure 1. Same strategies were applied to test the localization of 3C and 3D proteins, two other EV71 nonstructural proteins. However, overexpressed 3C and 3D proteins were dispersed in the cytoplasm (Fig. 2B, C). These results indicate that viral 2C protein plays an important role in the formation of IBs in vitro.
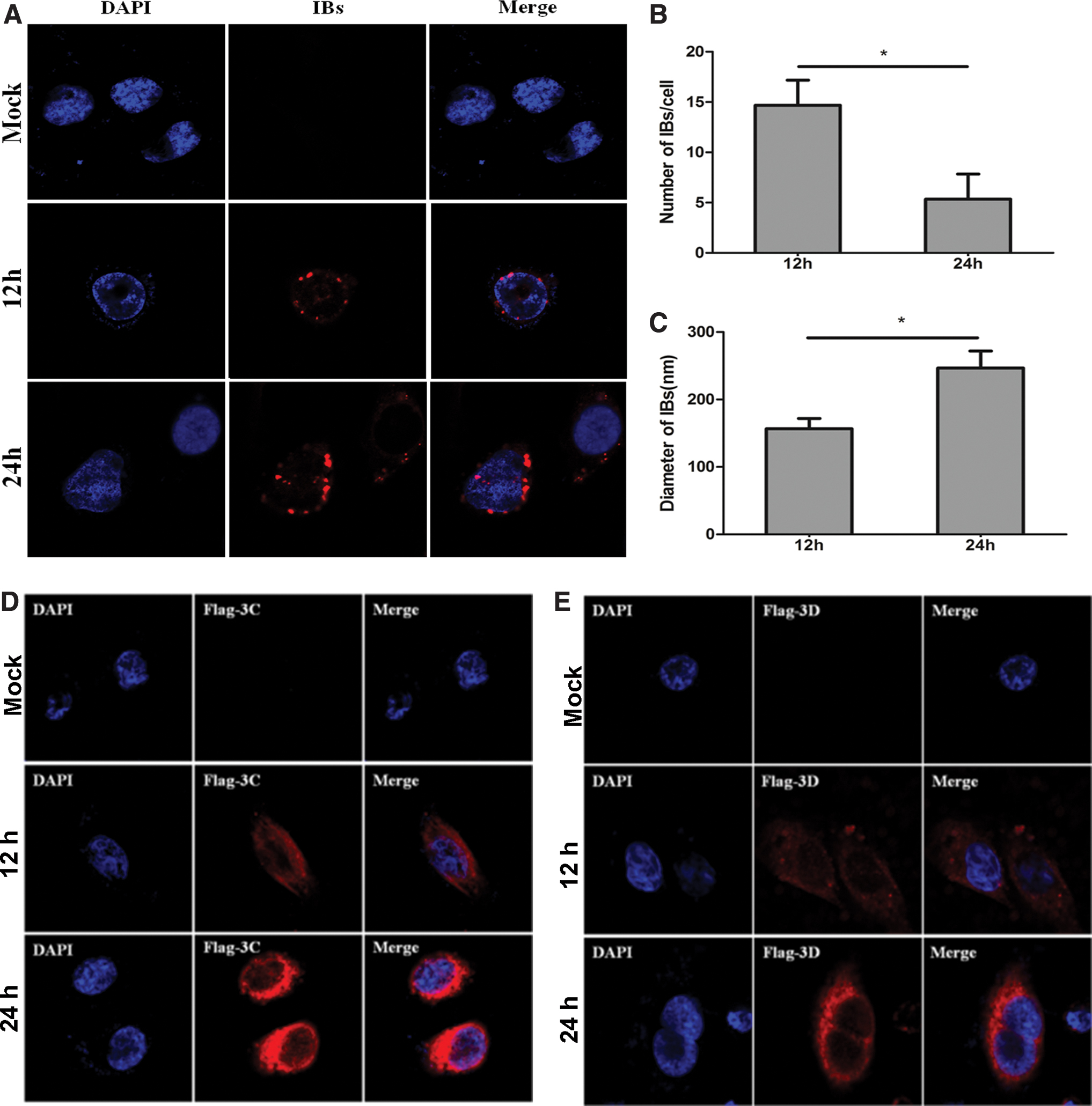
IBs were formed in 2C-transfected cells. HeLa cells were transfected with plasmids expressing HA-2C
IKKβ is translocated into IBs through direct interaction with 2C
Zheng et al., reported that EV71 2C protein interacts with IKKß by using coimmunoprecipitation (co-IP) and confocal immunofluorescence and 2C inhibits the phosphorylation of IKKß (22). We have confirmed the phenomenon in our Supplementary Figure S1 by using co-IP assay. We further explored the cellular localization of IKKβ in the presence of 2C since 2C protein was located in IBs as shown in Figures 1 and 2. As shown in Figure 3A, endogenous IKKβ formed foci and colocalized with 2C in the EV71-infected cells at the indicated time points. In contrast, IKKβ was dispersed in the cytoplasm in uninfected cells. In addition, in HeLa cells transfected with Flag-tagged IKKβ, IKKβ was dispersedly distributed in the cytoplasm (Fig. 3B). However, we observed that Flag-tagged IKKβ colocalized with 2C and IKKβ was recruited into IBs in the 2C-overexpressed cells (Fig. 3C). These results together indicate that sequestration of IKKβ into IBs occurred in the presence of 2C.
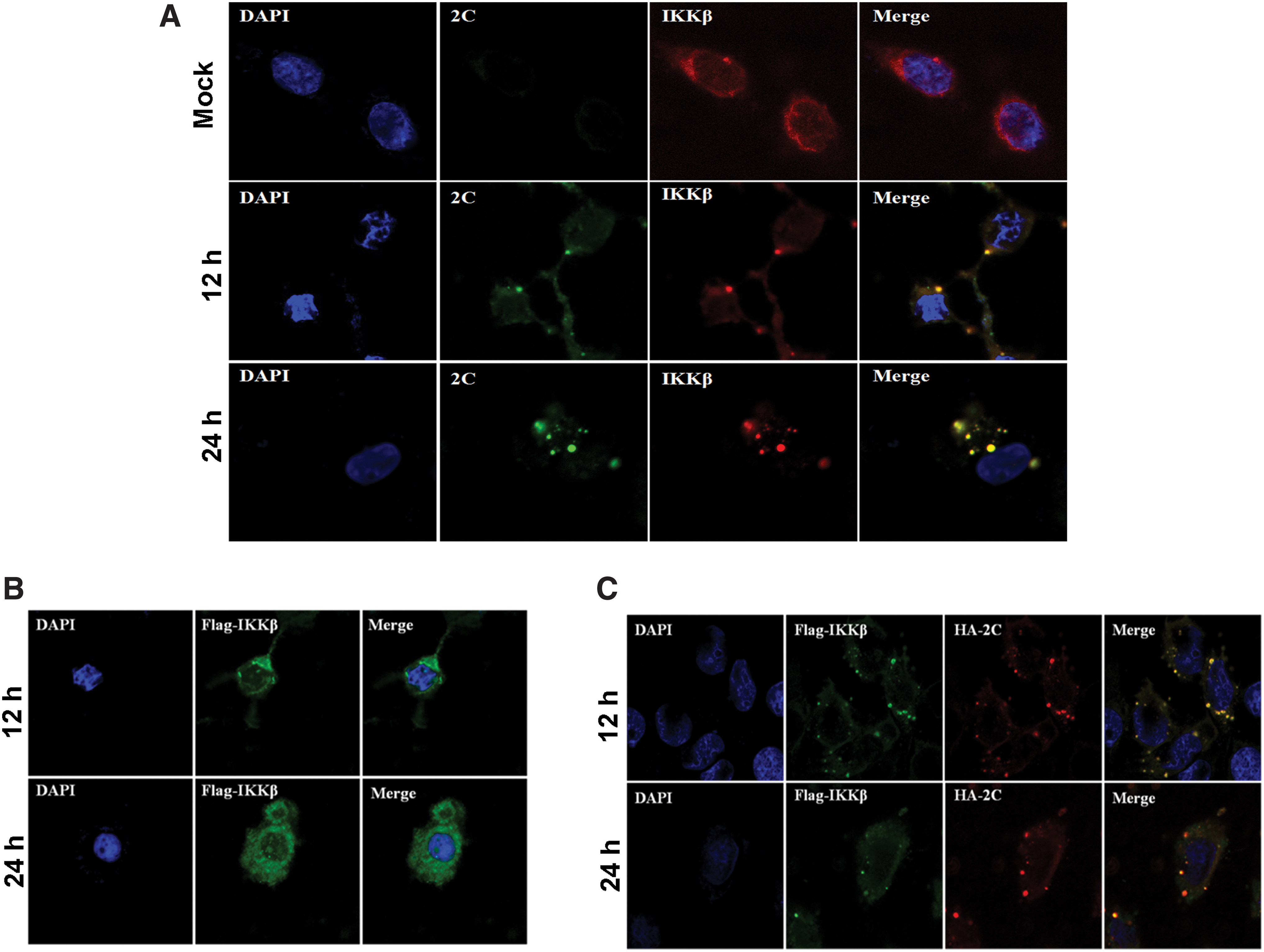
Colocalization of 2C with IKKβ through direct interaction in IBs.
IKKα is recruited into IBs through interaction with IKKβ
IKKα and IKKβ are two important members of IKK complex (6). In Supplementary Figure S1, we found 2C directly interacted with IKKβ but not IKKα. Since IKKα interacts with IKKβ (3), we questioned whether IKKα could be also trapped in IBs through interaction with IKKβ. To test this idea, we transfected HeLa cells to let overexpressing IKKα alone or IKKα together with IKKβ in the presence of 2C (Fig. 4). As shown in Figure 4A and B, IKKα was primarily located dispersedly in the cytosol in the presence or absence of IKKβ, respectively. Co-overexpression of IKKα and 2C did not bring IKKα into IBs (Fig. 4C), which was consistent with previous data, indicating that IKKα and 2C did not directly interact (Supplementary Fig. S1). However, when the cells co-overexpressed with IKKα, IKKβ, and 2C together, we found that IKKα colocalized in the IBs along with IKKβ and 2C (Fig. 4E). Based on the fluorescence intensity, it is likely that the expression of IKKα and IKKβ was not affected by 2C overexpression.

IKKα was translocated into IBs through interaction with IKKβ in the presence of 2C. HeLa cells were transfected with plasmids expressing Flag-IKKα alone
EV71 2C protein inhibits TNF-α-induced IKKα phosphorylation
A previous study has confirmed that 2C could directly interact with IKKβ and inhibit IKKβ phosphorylation (22). Although we found that 2C did not directly interact with IKKα (Supplementary Fig. S1), we questioned whether its phosphorylation state would be altered due to the colocalization with 2C in the IBs. HeLa cells stimulated with TNF-α (10 ng/mL) at the indicated time before transfecting with 2C-expressing plasmid. The phosphorylated IKKα in the cells transfected with 3 μg 2C-overexpressing plasmid was only a third of that transfected with an empty vector (Fig. 5A). Figure 5B shows the quantification results of Figure 5A, which shows TNF-α-induced IKKα phosphorylation was significantly suppressed by 2C at 1 μg (Fig. 5B). Taken together, these data indicated that 2C could suppress IKKα phosphorylation although they did not interact with each other.

(1) The effects of EV71 2C on TNF-α-mediated-IKKα activation.
EV71 2C protein suppresses IKKα phosphorylation with the help of IKKβ
We further investigated whether EV71 2C suppresses IKKα phosphorylation with the help of IKKβ. Therefore, we designed to evaluate whether IKKα phosphorylation would call for the help of IKKβ. We cotransfected IKKα-, IKKβ-, and 2C-expressing plasmids into 293T cells with varying IKKβ-expressing plasmid. The phosphorylation of IKKα was detected by immunoblot assay. We found that phospho-IKKα was reduced to a third of that transfected with empty vector in the presence of 2C and IKKβ (Fig. 5C, D). Taken together, these data indicated that IKKβ plays a crucial role in 2C suppressing IKKα phosphorylation.
Discussion
Studies in recent years have revealed that IB composition differs substantially, even between closely related viruses (13). IBs are also thought to be sites of viral replication and transcription during the process of virus replication (11) and play important roles in viral immune evasion. Prior studies have presented us that de novo RNA synthesis occurs in IBs formed by different viruses, such as Ebola virus, vesicular stomatitis virus, rabies virus, respiratory syncytial virus, and human metapneumovirus (1,7,8,10,12). Moreover, Wu et al., previously reported that NS protein of severe fever with thrombocytopenia syndrome could form viral IBs upon viral infection (19). In our study, we first discovered that EV71 infection forms viral IBs in the presence of 2C (Figs. 1, 2).
EV71 2C protein has been shown to involve in many crucial steps during the virus life cycle (15); 2C protein of EV71 interacts with viperin through its N-terminal domain, which is also the key domain responsible for the antiviral function of viperin (18). 2C protein has been functioned as an RNA helicase and ATP-independent RNA chaperone during EV71 infection (20). 2C binds to host-encoded Reticulon-3, who functions as a cellular modifier of enterovirus infection and replication (16). EV71 2C associates with IKKβ and p65 to inhibit NF-κB activation to escape from viral immunity (4,9,22). However, the details of 2C involvement in IB formation remain to be understood.
In our study, we found that 2C could inhibit IKKα phosphorylation in TNF-α-mediated NF-κB activation, although 2C did not directly interact with IKKα as evaluated by co-IP (Supplementary Fig. S1). The co-overexpression of IKKβ with 2C and IKKα in 293T cells reduced the phosphorylation level of IKKα, which indicated that IKKβ plays a crucial role in 2C suppressing IKKα phosphorylation.
In our study, we showed that 2C could form viral IBs in either infected or transfected cells. When HeLa and 293T cells were infected with EV71, 2C was able to form the unique structure of IBs in HeLa cells and 293T cells. However, viral protein 3C and 3D were dispersedly distributed in the cytoplasm in cells overexpressed with HA-3D or HA-3C, respectively. These results together suggested that 2C is specifically responsible for forming IBs in cells with EV71 infection.
We found that IBs formed by EV71 2C played a role in viral modulation against host immune responses. Our study suggested that EV71 2C could interact with IKKβ but not IKKα. IBs could sequester IKKβ due to their direct interaction between 2C and IKKβ. We actually have tried to figure out whether endogenous IKKβ is sufficient to translocate IKKα into IBs induced by 2C. We could not observe the weak signal of IKKα colocalized with 2C due to the low amount of endogenous IKKβ. That is why we used overexpressed IKKβ instead of endogenous IKKβ to magnify the observed IBs. Surprisingly, IKKα only moved into IBs in the presence of 2C together with IKKβ. In conclusion, 2C protein plays an important role in fighting against host immunity during EV71 infection. It sequestered IKKα and IKKβ into IBs and prevented their phosphorylation. This would inhibit host interferon beta (IFN-β) and NF-κB responses, which favors a more successful infection outcome. Since 2C serves as the primary bait in the IBs, we suggest that the 2C protein may be a potential therapeutic target to against EV71 infection. Here is what we think. (1) EV71 infection promoted the formation of IBs due to one of its nonstructural protein 2C. (2) 2C sequestered IKKβ into the IBs via direct interaction between them. (3) IKKα was translocated into IBs by interaction with IKKβ. (4) The phosphorylation of IKKα was reduced because it is sequestered in the IBs. (5) Diminished phosphorylated IKKα lead to reduced IFN-β and NF-κB responses, which favors a more successful EV71 infection outcome.
Footnotes
Acknowledgment
We are grateful to Xiaodong Wu, Institute of Microbiology, NJU, for technical help with confocal microscopy. We also thank Xiaolu Jiang for her assistance with the experiments. S.H. (Medical school of Nanjing University) revised the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by a research grant provided by National Natural Science Foundation of China (Grant No. 81672020 and Grant No. 81900823) and Science and Technology Development Fund of Nanjing Medical University (Grant No. NMUB2019213). This project was also supported by Nanjing Medical Science and Technology Development Fund (No.YKK19109).
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.