
Abstract
Human respiratory syncytial virus (RSV) is one of the major causes of childhood acute lower respiratory tract infection worldwide. Autophagy is an intracellular pathway involved in nutrient recycling. Recently, autophagy has been reported to play a role in regulating host cytokine response to several viruses, including vesicular stomatitis virus and human immunodeficiency virus. Previous in vivo studies using mouse model has shown that inhibition of autophagy reduces RSV-induced cytokine production. However, the role of autophagy in modulating RSV-induced cytokine response in human cells has not been reported. We investigated the role of autophagy in regulating the production of the cytokines C-X-C motif ligand 8 (CXCL8) and C-C motif ligand 5 (CCL5), in RSV-infected human bronchial epithelium BEAS-2B cells. Fluorescent microscopic analysis showed that RSV infection induced autophagosome formation in BEAS-2B cells. This autophagy inducing ability of RSV was further confirmed by flow cytometry. The effects of pharmacological inhibition of autophagy by SAR405 or chloroquine on cell death and cytokine release were quantified using lactate dehydrogenase assay and enzyme-linked immunosorbent assay (ELISA), respectively. We found that SAR405 or chloroquine did not cause cell death. Importantly, ELISA analysis showed that pharmacological inhibition of autophagy by SAR405 or chloroquine did not affect the productions of both CXCL5 and CXCL8. In contrast to the previous studies using mouse model, our data suggest that pharmacological inhibition of autophagy may not be a suitable strategy in controlling RSV-induced airway inflammation.
Introduction
Human respiratory syncytial virus (RSV) is a negative-sense single-stranded RNA virus which belongs to Pneumoviridae family (13). It is one of the leading causes of acute lower respiratory infection, which is associated with varying symptoms such as cough, difficulty in breathing, bronchiolitis, and severe pneumonia (3,6). RSV infection is responsible for significant morbidity and mortality among young children younger than 5 years old, the elderly, and individuals with chronic respiratory illnesses worldwide (3,10,29). Furthermore, hospitalization due to RSV in infancy has been associated with an increased risk for developing asthma and recurrent wheezing later in life (8,30). Despite years of effort, currently, there are neither licensed active prophylactic vaccines nor specific antiviral drugs against RSV (1,4,18). This is thought to be due to the complex nature of host and viral factors contributing to the disease pathogenesis, and the fact that natural infection provides limited protection from reinfection and disease (15). Thus, there is a need to better understand the regulations of RSV-induced host immune response that can limit viral replication, without causing immunopathogenicity to the host.
Airway epithelial cells have been shown to be the primary site of RSV infection (34). Following RSV infection, the innate immune response of airway epithelial cells is triggered upon the recognition of viral pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors called toll-like receptors (TLRs) and RIG-I-like receptors (RLRs). As a result of this recognition, a cascade of response is activated to induce the production and release of proinflammatory cytokines and antiviral interferons (IFNs) (27). However, the severity of RSV infection in susceptible individuals is largely driven by an overexuberant inflammatory response to the virus and is characterized by mucus hypersecretion and infiltration of neutrophils into the airways (32). Importantly, the C-X-C motif ligand 8 (CXCL8), a potent attractant of neutrophils, has been shown to contribute to acute airway inflammation following RSV infection (28). Therefore, a complete understanding of all the mechanisms that regulate cytokine production during RSV infection is crucial to further refine the therapeutic strategies to alleviate the excessive RSV-induced inflammatory response.
Recently, there is a growing interest in exploring whether autophagy plays a role in cytokine regulation (35). Autophagy is an intracellular, self-degradative process, which plays a major role in cell homeostasis maintenance and cell survival machinery following nutrient stress and pathogen invasion (7). Apart from mediating cell homeostasis maintenance and survival, autophagy has recently been reported to be involved in TLRs-mediated viral recognition by facilitating the delivery of cytosolic viral PAMPs to endosomal TLRs. Consequently, cytokine production is suppressed as a result of the autophagy inhibition (11,19).
Previous in vivo studies using mouse model has shown that inhibition of autophagy reduces RSV-induced cytokine production (17,21,24 –26). However, to our knowledge, the role of autophagy in modulating RSV-induced cytokine production in human cells has not been reported. Therefore, we sought to address the potential role of autophagy in regulating the cytokine responses of human lung epithelial cells to RSV infection.
Materials and Methods
Cell culture
IRB approval is not required for the use of commercially available (purchased) human cell lines. Immortalized human lung epithelial BEAS-2B cell line was purchased from American Type Culture Collection (ATCC, U.S.) and was cultured as described previously (9). In brief, the BEAS-2B cells were grown and maintained in Roswell Park Memorial Institute 1640 supplemented with 10% (v/v) fetal bovine serum and incubated in a humidified incubator at 37°C with 5% carbon dioxide (CO2) until the cells reached 80–90% confluency. The cells were routinely subcultured using Trypsin-EDTA (0.25%) with phenol red (Gibco).
Virus propagation and titration
Human RSV antigenic subgroup B strain 18537 was purchased from ATCC, U.S., and propagated in HEp-2 cervical cancer cells (HeLa contaminants; ATCC CCL-23) until 60–70% of cytopathic effects were observed as described previously (36). Cell lysates containing the virus were then collected via centrifugation at 1,008 g for 7 min at 4°C. Viral titer was determined by plaque assay using HEp-2 cells as described previously (16).
Cell infection and treatment with pharmacological autophagy inducers
BEAS-2B cells were seeded (30 × 104 cells/wells for fluorescence microscopy and 50 × 104 cells/wells) for flow cytometry) in 24-well plate. On the following day, the cells were left uninfected (mock control) or infected with RSV at multiplicity of infection (MOI) of 1 and allowed for adsorption for 2 h in a humidified incubator. The remaining viral inoculum was removed, and infected cells were then incubated for 24 h. Meanwhile, uninfected cells were treated with the autophagy inducer Torin2 (20 nM) for 24 h. The ability of RSV in inducing autophagy was then examined using fluorescence microscopy and flow cytometry.
Fluorescence microscopy and flow cytometry
Autophagy induction in RSV-infected BEAS-2B cells was qualitatively visualized by Cyto-ID® Autophagy Detection Kit-based fluorescence microscopy, following manufacturer's instruction manual. In brief, the cells were first washed with phosphate-buffered saline before being stained with 100 μL of solution containing 0.2 μL Cyto-ID Green stain for 40 min in the dark. Green fluorescence signals indicating the presence of autophagosomes were visualized under Eclipse Ti2 fluorescent microscope (Nikon, Japan) using 20 × Extra Long Working Distance (ELWD) lens. The autophagy induction level was then quantified using flow cytometry as described in manufacturer's manual. In brief, the cells were trypsinized and centrifuged at 252 g for 5 min. The cell pellets were subsequently resuspended in 200 μL of solution containing 0.4 μL Cyto-ID Green stain for 30 min in the dark before being analyzed using flow cytometry.
Cell infection and treatment with pharmacological autophagy inhibitors
BEAS-2B cells were left uninfected (mock control) or infected with RSV at MOI of 1 and allowed for adsorption for 2 h in a humidified incubator. The unbound viral inoculum was then removed, and the cells were either left untreated or treated with the autophagy inhibitor SAR405 (200 nM) or chloroquine (2 μM). After 48 h incubation, the supernatants and cell lysates were harvested for further assays.
Lactate dehydrogenase cytotoxic assay
Lactate dehydrogenase (LDH) assay was performed to quantify cell death. Before performing the assay, cell-free supernatants and cell lysates were harvested as described previously (9). Fifty microliters of the original cell-free supernatants or media of freeze-thaw cell lysis was added into the wells of a 96-well plate. Next, 50 μL of substrate reagent was added into each well and incubated for 30 min in the dark. Fifty microliters of stop solution was then added into each well and the absorbance was immediately measured using a microplate reader (Bio-Rad) at 490 nm wavelength.
Enzyme-linked immunosorbent assay
Secreted cytokine concentrations in cell-free supernatants were quantified by enzyme-linked immunosorbent assay (ELISA) in a 96-well plate as described in manufacturers' manuals. Concentrations of CXCL8 and C-C motif ligand 5 (CCL5) were measured using matched antibody pairs from R&D System and Invitrogen, respectively. Absorbance was measured using a microplate reader (Bio-Rad) at 450 nm wavelength.
Statistical analyses
Data are presented as mean ± standard deviation. Statistical comparisons were performed using GraphPad Prism v6.0 software, using the indicated tests.
Results
RSV infection induces autophagy in lung epithelial cells
Previous studies have shown that RSV can induce autophagy in human cervical cancer cells (13) and human fibrosarcoma cells (31). However, the ability of RSV in inducing autophagy in human lung epithelial cells, which are the main site of infection for RSV has not been reported. To assess autophagy induction in BEAS-2B human lung epithelial cells, we used Cyto-ID Autophagy Detection Kit, which contains a novel dye that selectively labels autophagosomes. Using fluorescence microscopy, it was observed that RSV remarkably induced autophagosome formation in BEAS-2B cells compared to mock, at 24 hours post infection (h.p.i.) (Fig. 1A). We then further confirmed this qualitative data using flow cytometry analysis. Similarly, there was an increase of autophagic flux in BEAS-2B cells following RSV infection at 24 h.p.i. (Fig. 1B).

RSV induces autophagy in lung epithelial cells. BEAS-2B cells were left untreated/uninfected (mock), or treated with autophagy inducer, Torin2 (positive control), or infected with RSV at MOI = 1. At 24 h.p.i., cells were stained with Cyto-ID Autophagy Detection Kit.
Cell viability of lung epithelial cells is not affected by autophagy inhibition
To investigate the effects of autophagy inhibition on the lung epithelial cell responses to RSV, we used the pharmacological autophagy inhibitors SAR405 and chloroquine. SAR405 inhibits autophagy at the early stage, while chloroquine is a late-stage autophagy inhibitor (22,23). Before measuring the effect of autophagy inhibition on cytokine production, we first sought to confirm that both SAR405 and chloroquine at the optimized concentrations do not cause cell death. After being infected with RSV, we treated the cells with the optimal concentration of SAR405 or chloroquine for 48 h. Based on our microscopic observation, the morphology of BEAS-2B lung epithelial cells was not changed following treatment with SAR405 or chloroquine (Fig. 2A). We further quantified this microscopic observation of cell death using LDH cytotoxic assay. After 48 h treatment, it was found that both SAR405 and chloroquine did not cause toxicity to the BEAS-2B cells as they only resulted in <5% cell death, similar to that of untreated control (Fig. 2B). However, both autophagy inhibitors slightly caused cell death in RSV-infected BEAS-2B cells, although they were not significant (Fig. 2B).
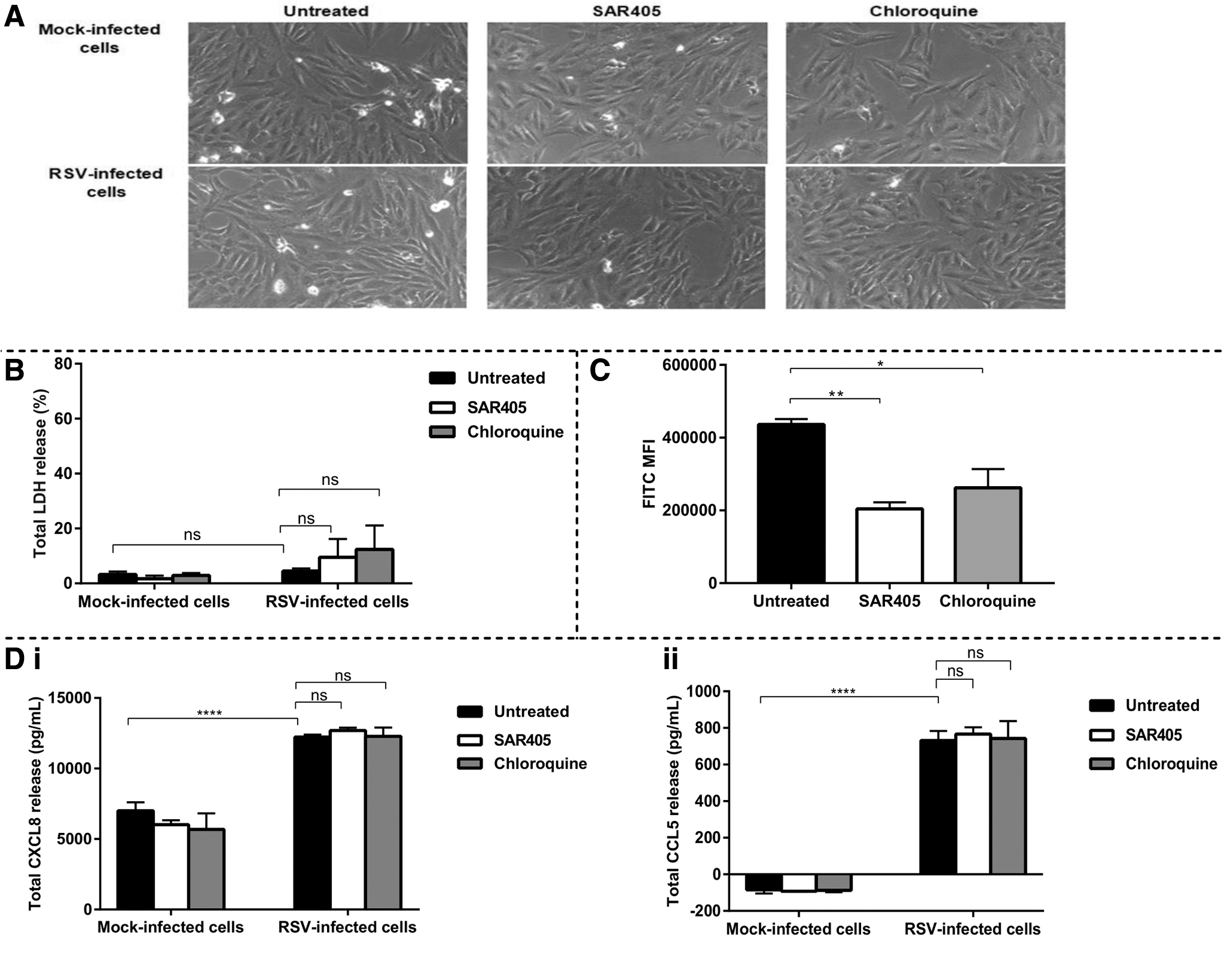
(
SAR405 and chloroquine inhibit RSV-induced autophagy in lung epithelial cells
To verify the ability of SAR405 and chloroquine to inhibit autophagy in BEAS-2B cells during RSV infection, we treated BEAS-2B cells with SAR405 and chloroquine following infection with RSV. Cells were incubated for 90 min before being harvested and stained with Cyto-ID Autophagy Detection Kit. Subsequently, flow cytometry analysis was performed to measure the autophagic flux in BEAS-2B cells. Based on the flow cytometry profiling, we found that both SAR405 and chloroquine significantly inhibited RSV-induced autophagy in the BEAS-2B lung epithelial cells (Fig. 2C).
Autophagy does not regulate cytokine production in lung epithelial cells during RSV infection
After confirming that both SAR405 and chloroquine do inhibit autophagy in RSV-infected BEAS-2B cells, we further investigated the effect of autophagy inhibition on cytokine production in the BEAS-2B cells. The release of the chemokines CXCL8 and CCL5 following RSV infection was determined by ELISA. CXCL8 is a potent neutrophil attractant, and neutrophil infiltration is strongly linked to disease severity caused by RSV infection (5). CCL5, which is an IFN-stimulated gene (ISG) product is a chemoattractant for eosinophils, and eosinophilic inflammation has been shown to be detrimental in RSV-infected patients (2, 20). As expected, RSV infection triggered significant releases of CXCL8 (Fig. 2D(i)) and CCL5 (Fig. 2D(ii)) in BEAS-2B cells, as measured at 48 h.p.i. However, treatment with the pharmacological autophagy inhibitor SAR405 or chloroquine neither elevated nor inhibited the productions of CCL5 and CXCL8 in the RSV-infected lung epithelial cells.
Discussion
Human RSV remains an important viral pathogen as it is the leading cause of acute lower respiratory infection in children younger than 5 years old (33). In this study, we show that autophagy inhibition does not affect cytokine responses in human lung epithelial cells following RSV infection.
In recent years, there is a growing interest in investigating whether autophagy plays a role in cytokine regulation (35). Previous in vivo studies using mouse model has demonstrated that autophagy inhibition reduces RSV-induced cytokine production (17,21,24 –26). However, the role of autophagy in modulating RSV-induced cytokine production in human cells remains unknown. To elucidate this, we first examined the effects of RSV infection on autophagy in human lung epithelial cells. We observed that RSV could induce autophagy in BEAS-2B lung epithelial cells, in keeping with previous published data using other human cell types (13, 36).
To test our hypothesis on the potential effect of autophagy inhibition on cytokine production, we further treated the RSV-infected cells with the autophagy inhibitors SAR405 and chloroquine. SAR405 is an early stage autophagy inhibitor, which interrupts the formation of class III PI3-kinase (PI3KC3) complex, while chloroquine is a late stage autophagy inhibitor, which inhibits autophagosome-lysosome fusion (12,14,22). Before measuring the cytokine release, we sought to confirm that both SAR405 and chloroquine at the optimized concentrations did not cause cell death. This study is the first to demonstrate that both compounds SAR405 and chloroquine are not toxic to RSV-infected human lung epithelial cells. Our data are not consistent with the previous study using another known autophagy inhibitor, 3-methyadenine (3-MA), which showed that 3-MA caused significant cell death in RSV-infected human cervical cancer cells as measured at 48 h p.i (13). Whether 3-MA can also cause cell death in human lung epithelial cells will require further investigation. Furthermore, before measuring the cytokine production, we also confirmed that both SAR405 and chloroquine did inhibit autophagy in the RSV-infected BEAS-2B lung epithelial cells.
We then determined if autophagy plays a role in cytokine regulation in human lung epithelial cells. In contrast to the previous studies using mouse model (17,21,24 –26), we found that pharmacological inhibition of autophagy did not affect innate cytokine production in human lung epithelial cells during RSV infection. However, in the present study, we measured CXCL8, while none of the published mice studies quantified CXCL8 production. CXCL8 is a chemoattractant cytokine that primarily recruits neutrophils, and neutrophilic inflammation is a major contributor of disease severity caused by RSV infection (20, 28). Apart from CXCL8, we also measured CCL5 production. CCL5 is a chemokine for eosinophils, and eosinophilic inflammation has been shown to contribute to airways damage in RSV-infected patients (2,20). We found that RSV-induced CCL5 production in human lung epithelial cells was not affected by inhibition of autophagy using SAR405 or chloroquine, in disagreement with the previous mice studies (17,21). These previous studies, however, used 3-MA, and again, it is not known if the discrepancy of findings was due to different pharmacological inhibitors used. Interestingly, a study using knockout mice lacking the autophagy protein Beclin-1 has reported that following RSV infection, the production of CCL5, and several other innate cytokines was reduced in pulmonary dendritic cells, but remained unaffected in lung epithelial cells (25). This suggests that autophagy may pose different roles in different cell types.
One major limitation of our study is the fact that autophagic activity was not completely inhibited by the pharmacological inhibitors SAR405 and chloroquine. Thus, future studies using other approaches to inhibit autophagy such as short-interfering RNA-mediated knockdown of autophagy proteins are needed to confirm our current findings obtained using pharmacological inhibitors.
Overall, our data showed that RSV infection induces autophagy in human lung epithelial cells. Importantly, we reported for the first time that inhibition of autophagy by pharmacological inhibitors does not affect RSV-induced innate cytokine production in human lung epithelial cells. Again, further studies using other approaches to inhibit autophagy will validate if targeting autophagy is not a suitable strategy to alleviate RSV-induced airway inflammation.
Footnotes
Acknowledgment
The authors thank Dr. Sze Wei Leong for the technical assistance in performing flow cytometry.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research has been funded by Universiti Putra Malaysia's GP-IPM grant (9544600) and GP-IPS grant (9669100).