
Abstract
Herpes simplex virus (HSV)-1 infection causes cold sores and keratitis. Upon infection, it forms lesions at the epithelium and enters neurons where it establishes a latent infection. Host innate immune receptor Toll-like receptor (TLR)2 recognizes HSV by sensing its glycoproteins and induces an innate immune response. Upon activation, TLR2 forms a dimer with TLR1, TLR2, or TLR6 and signals inducing cytokines and interferons (IFNs). In this study, we checked the effect of differential activation of TLR2 by using different TLR2 dimer-specific ligands on the anti-HSV-1 innate immune response. We found that TLR2/2 ligand-induced IFN-β in neurons, while IFN-α in astrocytes and these IFNs subsequently induce the expression of IFN stimulatory genes like viperin, Ch25H, OAS2, latent RNase (RNase L), protein kinase R (PKR), and interferon-induced proteins with tetratricopeptide repeats (IFIT) 1. These are the genes with antiviral functions such as blocking viral attachment, protein synthesis, and egress.
Introduction
Herpes Simplex Virus 1 (HSV-1) is a causative agent of cold sores, herpes keratitis, and encephalitis. It infects at the epithelial surfaces forming lesions and subsequently enters into sensory neurons and establishes latent infection in trigeminal ganglia. During the course of infection, HSV-1 is detected by the host-pathogen recognition receptors like Toll-like receptors (TLRs). It is shown that the TLR2 recognizes viral glycoproteins like HSV-gB, HSV-gD, and HSV-gH-gL, while TLR9 recognizes the viral double-stranded DNA (dsDNA) genome. Studies have revealed the protective role of TLR2, TLR3, and TLR9 during HSV infection, but some studies reported exacerbative functions of TLR2 in encephalitis caused by HSV-1 infection (3,15,22,23,38,48,52).
During HSV infection at the genital or orofacial surfaces, TLR2 and TLR9 in the macrophages, dendritic cells (DC), and epithelial cells recognize the viral envelope proteins and CpG DNA, respectively (41,52). Both TLRs are expressed in the trigeminal ganglion and work synergistically, inducing inducible nitric oxide synthase (iNOS), which is critical against HSV infection (48). TLR2 has been found to mediate proinflammatory immune response by secreting chemokines (CCL 7, 8, and 9 and CXCL 1, 2, 4, and 5) and cytokines (TNF-α, interleukin [IL]-1β, IL-6, and IL-12) by microglial cells in response to HSV-1 (3). At the time of infection, gH/gL and gB interact with TLR2 and activate the NF-κB signaling pathway, resulting in the secretion of IL-6 and interferon (IFN)-β, which are known to have an anti-HSV function (15,23). Apart from this, NK cells are found to be activated after HSV infection by TLR2 signaling. NK cells play a role in antigen (Ag) presentation to CD4+ T cells or assist DCs in Ag presentation (20). Stimulation of TLR2 on regulatory T (T-reg) cells reversed its regulatory function and thus promoted the protective function of HSV-specific effector CD4+CD25+ T cells (33). In addition to HSV surface proteins, fibroblast-stimulating lipopeptide-1 (FSL-1), a TLR2/6 agonist, enhanced TLR2-mediated resistance to HSV-2 infection (39). Recently, it is reported that a self-adjuvanting genital tract peptide-lipid conjugate vaccine elicited local and systemic CD8+ T cell response and protected the mice from HSV-2 challenge (51). In addition, myeloid-derived dendritic cells recognize and respond to the HSV-1 through 4 glycoproteins gB, gD, gH, and gL the ligands for virus entry into the cell, irrespective of other protein and nucleic acid recognition by TLR2 (23).
TLRs are involved in the protective immune response against HSV [10], but the microglial cells in TLR2 knockout mice were found to have reduced apoptosis and deaths 24 h after HSV-1 infection and had the downregulated expression of proapoptotic genes (2). Kurt-Jones et al. showed that TLR2 contributed to lethal encephalitis during HSV-1 infection by enhancing MCP-1 expression and inflammation in the brain and related the mortality to TLR2 activation and not to the HSV-1 level in mouse brain (22). These findings were supported by the observations that TLR2 on microglia induces an inflammatory response, reactive oxygen species (ROS), and oxidative stress leading to neuronal damage after HSV infection (18,52).
The above findings, which implicate the protective as well as the damaging role of TLR2, fall short of generating a clear idea about the role of TLR2 during HSV infection. Following activation, TLR2 dimerizes with TLR1, TLR2, or TLR6 forming TLR2/2 homodimer, or TLR1/2 or TLR2/6 heterodimer, respectively, and activates NF-κB, interferon regulatory factor (IRF)-3 and induces IFN and cytokines (4,19). It is not reported whether TLR1/2, TLR2/2, or TLR2/6 dimerization plays selective roles during HSV infection. HSV-1 infects and establishes latent infection in neuronal cells, while astrocytes are also infected by HSV-1. We, therefore, studied the signaling through TLR2 dimers in HSV-1F-infected neuronal cell line Neuro2a and astrocyte cell line DBT by selectively inducing its activation by using specific TLR2 ligands. Upon infection and TLR2 ligand stimulation, we checked the expression of IFNs and interferon-stimulated genes (ISGs), which have direct or indirect antiviral functions.
Materials and Methods
Cell cultures and viral plaque assay
Vero cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and infected with HSV-1F after it reached confluency. Cells were incubated until the virus established infection, as observed by the cytopathic effect or detachment of cells. These cells with whole media were harvested centrifuged at 3,220 × g to separate the debris. The supernatant was then centrifuged at 56,000 × g at 4°C for 2 h, the medium was discarded, and the pellet was resuspended in the phosphate-buffered saline. The viral concentration (PFU/mL) was determined by plaque assay. Vero cells were plated in a 12-well plate and incubated till it reaches confluency. The diluted virus stock was used to infect the Vero cells (dilution up to 10−8) for 90 min and overlaid with 2% agarose in 2 × media. The plaques were counted 48 h after incubation; PFU/mL was calculated to determine the virus number.
Neuroglial cell culture and virus infection assay
Astrocyte cell line DBT and neuronal cell line Neuro-2a were cultured in DMEM with 10% FBS (GIBCO) and Penicillin-Streptomycin (100 μg/mL) at 37°C in 5% CO2. In a 12-well plate, 2 × 105 cells/well were infected with HSV-1F multiplicity of infection (MOI)-1 and 0.1, respectively, and stimulated with different TLR2 ligands in DMEM for 8 h. The experimental set included uninfected cell control, infection control, ligand stimulation alone, and infection with ligand stimulation. Concentrations of ligands used for stimulation of neuron and astrocyte are TLR1/2 (Pam3csk4, 0.5 ng/mL), TLR2/2 (peptidoglycan [PGN], 7.5 μg/mL), and TLR2/6 (FSL-1, 1 ng/mL). Ligand concentrations for this study were determined by concentration-dependent neuron and astrocyte viability assay by MTT (Fig. 1A–D) and concentration-dependent expression of proinflammatory cytokine IL-12 in neurons (Fig. 1E–G) and astrocytes (Fig. 1H–J). MOI for infection was determined by MOI-dependent IL-12 expression in neurons and astrocytes (Fig. 1K, L).
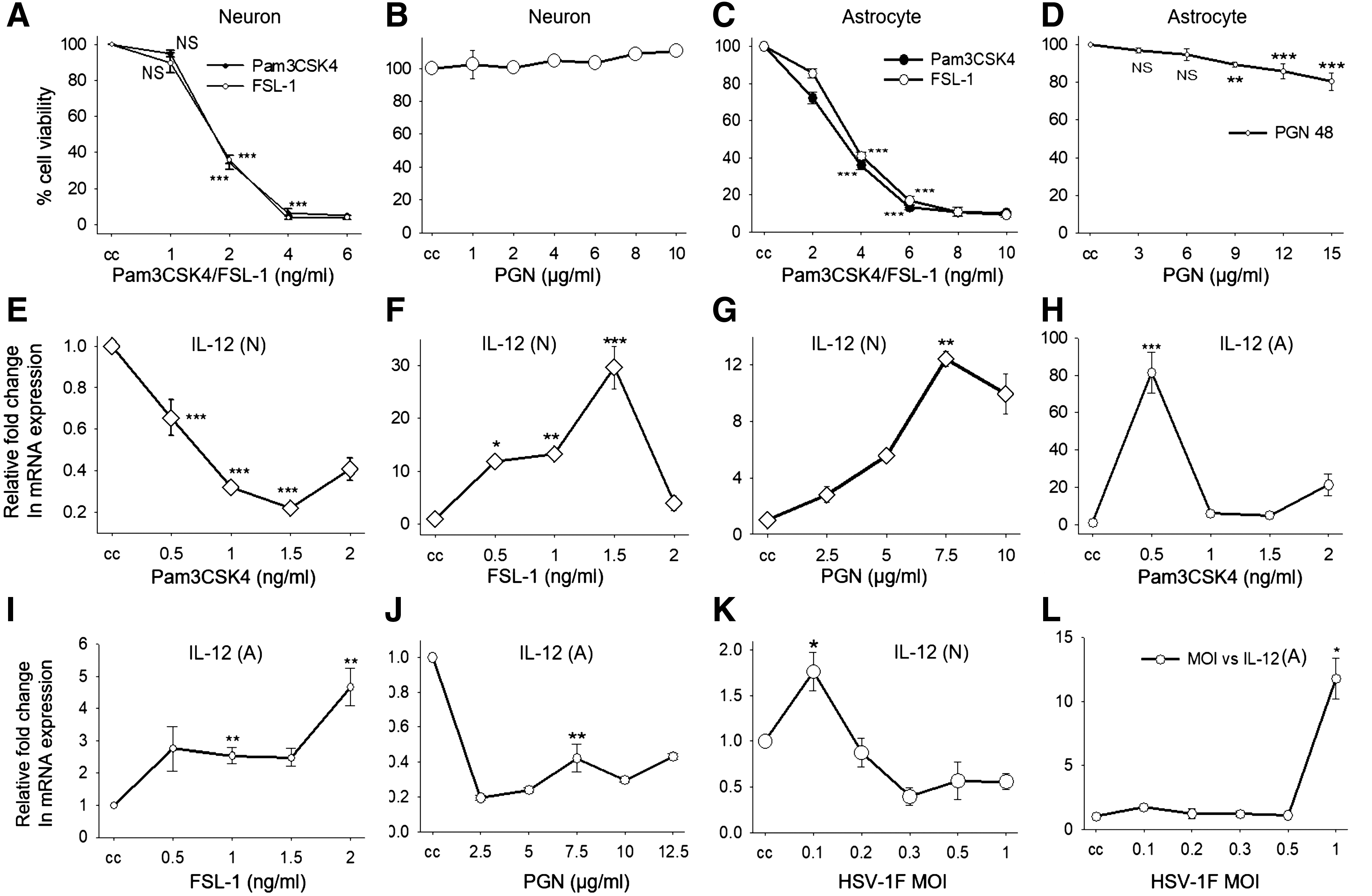
Cell viability assay by MTT
Neuronal (Neuro-2a) and astrocyte (DBT) cell lines were seeded in a 96-well plate with density of 104 cells/well. After 24 h, cells were treated with different concentrations of Pam3CSK4, FSL-1 and PGN for 48 h. Media containing ligands were removed and replaced with the media containing MTT reagent with a final concentration 0.5 mg/mL for 2 h. The medium was then removed and dimethyl sulfoxide (DMSO) was added to the cells and mixed well. The absorbance was measured spectrophotometrically at 570 nm and plotted as percent cell viability compared to untreated cell control.
RNA isolation, complementary DNA preparation, and qRT-PCR
RNA was isolated from the infected/stimulated cells using Tri reagent. RNA was checked for quantity and quality by measuring the absorbance at 260/280 nm using a nanodrop spectrophotometer. Complementary DNA (cDNA) was prepared using RNA (2 μg), which was used as a template for gene expression analysis by quantitative real-time polymerase chain reaction (qRT-PCR). SYBR green fluorescent dye was used to check the real-time amplification, the program for PCR was Step 1: initial denaturation 95°C 30 sec, Step 2: 45 cycles (denaturation at 95°C for 5 sec and annealing and amplification at 60°C for 35 sec), and Step 3: melt curve. GAPDH was used as a control gene to calculate the relative fold change in the gene expression compared to unstimulated/noninfected control. The primers used in this study are given in Table 1.
Primers Used in the Study
IFIT, interferon-induced proteins with tetratricopeptide repeats; IFN, interferon; IL, interleukin; ISG, interferon-stimulated Gene; Mx-1, Myxovirus resistance 1; PKR, protein kinase R; TLR, Toll-like receptor.
Statistical analysis
Statistical analysis was done using Students t-test for cell viability assay, concentration- and MOI-dependent gene expression. One-way analysis of variance (Student-Newman-Keuls Method) was employed to analyze ligand-dependent innate response (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
Results
HSV-1 infection alters TLR expression in neurons and astrocytes
HSV-1 infects epithelial cells and causes lesions at infection site due to lytic infection. At this stage of infection, HSV-1 infects nearby sensory neurons and travels by retrograde transport up the axons to the trigeminal ganglia, where it establishes a latent infection. Apart from neurons, HSV-1 also infects astrocytes, oligodendrocytes, microglia specialized endothelial cells, and pericytes within the central nervous system (CNS) (6,12,25). Neurons and astrocytes recognize HSV-1 by sensing its pathogen-associated molecular patterns (PAMPs) like glycoproteins, single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), and dsDNA genome through different TLRs and RNA and DNA sensors during the course of infection (5,31). Among these PAMPs, HSV-1 glycoproteins gB, gH, and gL are reported to be sensed by TLR2 (23). The aim of this study was therefore to analyze selective signaling through TLR2 following its ligand recognition by forming homodimer with itself or heterodimers with TLR1 or TLR6. The recognition leads to activation of NF-κB and IRF-3, which translocate to the nucleus and induces expression of IFNs and ISGs. Signals from TLR2/2, TLR1/2, and TLR2/6 dimers may lead to differential IFN and ISG expression, but their effects leading to differential outcome of HSV pathogenesis remain unknown. These selective effects might result from differential NF-κB activation by a particular dimer or abundance of a particular PAMP on HSV facilitating selective dimerization or HSV-1-induced selective modulation of particular TLR expression.
The ligand concentrations used to trigger different TLR dimers were determined by concentration-dependent cell viability and IL-12 expression (Fig. 1A–J) in neurons and astrocytes. MOI for infection was determined by MOI-dependent IL-12 expression in neurons and astrocytes, as described in Materials and Methods section (Fig. 1K, L). For examining the effect of selective sensing of HSV-1 by different TLR dimers on the anti-HSV response, we checked the expression of TLR1, TLR2, and TLR6 on HSV-1F-infected neuronal and astrocyte cell lines. We observed that expression of TLR1, TLR2, and TLR6 was reduced in neurons, while in astrocytes, TLR1 and TLR6 were unaltered and TLR2 was reduced after HSV-1F infection (Fig. 2A, B). As TLR2 forms dimer with TLR1 or TLR6 for signaling, its reduced expression might have an impact on signaling through TLR1 and TLR6 in astrocytes. In astrocytes, TLR2/TLR2 dimer-induced signaling might be important for anti-HSV-1 response and suppression of TLR2 might be a viral invasion strategy. In neurons, the expression of all three TLRs was reduced, implying either their collective role in antiviral response or an HSV-1-adopted immune evasion strategy.
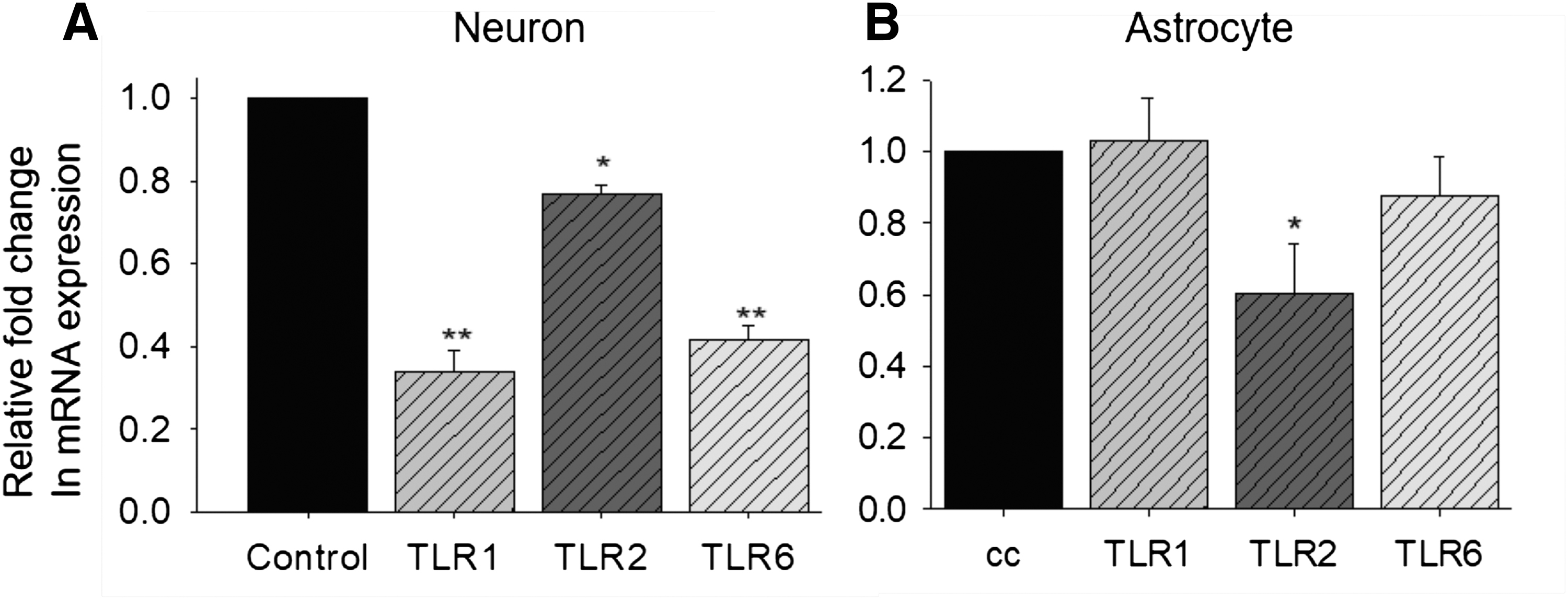
Neuronal and astrocyte cells were infected with HSV-1F and TLR1, TLR2, and TLR6 expression were checked using qRT-PCR in neurons
TLR2 dimers induce differential type-I IFN expression after HSV-1F infection
TLR2 recognizes viral glycoproteins, dimerizes, and recruits TIRAP and MyD88 to activate IRAK1, IRAK4, and TRAF6, which in turn lead to activation and translocation of the transcription factors NF-κB and IRF-3 to the nucleus, wherein it induces the gene coding for the cytokines TNF-α, IL-6, and IFN-α/β (3,19,23). We used synthetic ligands to activate signaling through particular dimers of TLR2: Pam3csk4 for TLR1-TLR2, PGN for TLR2-TLR2, and FSL-1 for TLR2-TLR6. We stimulated neuronal cell line Neuro2a and astrocyte cell line DBT with the indicated concentrations of these ligands for 8 h and checked type-I IFN expression profile in infected and uninfected cells by quantitative real-time PCR. We observed that IFN-α expression was reduced, while IFN-β and IFN-ɛ expression remained unaltered after infection in neurons. IFN-α was downregulated after TLR2 activations from all dimers compared to uninfected cell control (Fig. 3A). When TLR-stimulated neurons were infected with HSV-1F, there was no significant difference in IFN-α expression after infection (Fig. 3A). IFN-β was upregulated upon TLR1/2 and TLR2/6 stimulation and expression levels remained unchanged after infection. In TLR2/2-stimulated neuronal cells, IFN-β expression was increased after infection (Fig. 3B). IFN-ɛ expression was reduced after TLR2/2 and TLR2/6 stimulation in neurons, but it attained normal cellular levels after infection (Fig. 3C). In astrocytes, IFN-α expression was upregulated after infection (Fig. 3D). It is significantly increased when TLR2/2-stimulated cells were infected with HSV-1F. IFN-β expression was reduced upon infection and TLR2 stimulation with all three ligands (Fig. 3E). IFN-κ was undetected in neurons, but was reduced in astrocytes upon PGN treatment; by contrast, HSV-1 infection upregulated its expression (Fig. 3F). IFN-ɛ was reduced when TLR1/2-stimulated cells were infected. Upon TLR2/2 and TLR2/6 stimulation, its expression was reduced, but increased in TLR2/2-stimulated astrocytes after infection (Fig. 3G). IFN-α, a type-I IFN expression, is different in neurons and astrocytes after HSV-1F infection as IFN-α was reduced in neurons, while increased in astrocytes, while other IFN expression was unaltered after infection, suggesting HSV-1-induced IFN-α suppression in neurons and HSV-1-induced IFN-β suppression in astrocytes. TLR1/2 dimer stimulation with Pam3CSK4 increased IFN-β expression, while it decreased IFN-α in neurons after infection, suggesting its protective role in antiviral IFN response, particularly through induction of IFN-β. In astrocytes, TLR1-TLR2 induces IFN-α, while it represses IFN-β, thus inducing the antiviral IFN response through IFN-α. FSL-1-induced TLR2-TLR6 dimerization increased IFN-β expression, but reduced IFN-α in neurons, whereas IFN-α increased, but IFN-β reduced in astrocytes, suggesting that TLR2-TLR6 stimulation leads to IFN-α-dominated antiviral response in astrocytes, whereas through IFN-β in neurons. TLR2-TLR2 stimulation with PGN increases expression of IFN-β in neurons while IFN-α, IFN-κ, and IFN-ɛ were increased in astrocytes. These observations suggest that TLR2-TLR2 mediates IFN response through IFN-β in neurons, while it is a collective effect of IFN-α, IFN-κ, and IFN-ɛ in astrocytes.
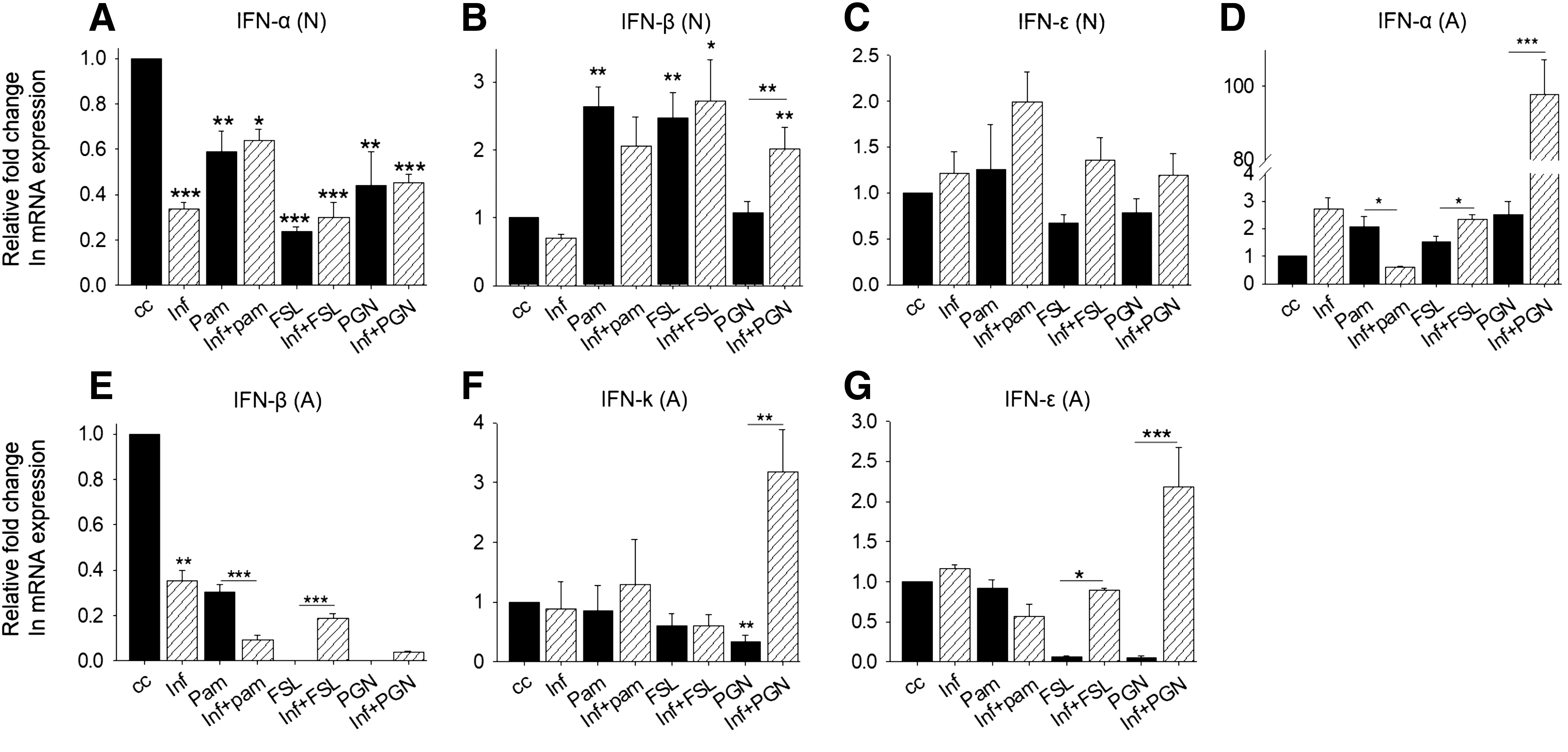
Type I IFN response in TLR2-stimulated neurons and astrocytes. Neuronal and astrocyte cells were treated with TLR2 ligand and infected with HSV-1F for 8 h and mRNA expression was checked by qRT-PCR. Plotted as relative fold change in mRNA expression (Y-axis) in given treatment/infection (X-axis) compared to untreated control. IFN expression after different treatments/infection in neurons
TLR2 dimers induce differential ISG expression in HSV-1-infected neurons and astrocytes
Type I IFNs bind to their receptors causing Janus Kinase (JAK) 1 and Tyrosine kinase 2 (Tyk2) phosphorylation and recruitment and phosphorylation of signal transducer and activator of transcription (STAT) 1 and STAT2. STAT1/2 heterodimer binds with IRF-9 forming ISGF3, which translocates to the nucleus and binds to ISRE and GAS elements in the promoters of ISGs, inducing their expression (37). ISGs are a group of proteins, induced by IFNs that have direct or indirect antiviral effects. ISGs mediate their functions by inhibiting viral transcription, protein synthesis, viral assembly-maturation, and cell to cell viral transmission (40). We checked the expression of ISGs like ISG15, viperin, protein kinase R (PKR), myxovirus resistance 1 (Mx-1), Ch25H, OAS2, OAS3, latent RNase (RNase L), and interferon-induced proteins with tetratricopeptide repeats (IFIT) 1–3 in neurons and astrocytes by qRT-PCR after treatment with TLR2 ligands and infection with HSV-1F.
TLR2 dimer induces viperin in neurons, while Ch25H in astrocytes
Lipid biosynthesis and membrane raft stability are affected by viperin and Ch25H. HSV is an enveloped virus and it needs a host cell membrane to make progeny viruses. Viperin controls the membrane fluidity and hence the viral egress (29,34,42). Ch25H enzyme converts cholesterol into 25HC that mediates suppression of SREBP activation and degradation of HMGCR and reduced cholesterol accessibility due to esterification-led localization to the core of lipid droplets. These events increase the membrane rigidity, reducing viral entry into host cells (26,45). Viperin expression was increased in Pam3CSK4-treated, that is, TLR1/2-stimulated neurons (Fig. 4A), while it is also increased in PGN-treated, that is, TLR2/2-treated astrocytes after HSV-1 infection (Fig. 4B). TLR2 ligand stimulation reduced Ch25H expression, compared to control, in neurons and astrocytes (Fig. 4C, D). In astrocytes, it was increased after TLR2/2 stimulation, but reduced significantly after HSV-1 infection. It suggests that neuronal TLR1/2 and astrocytic TLR2/2 signals contribute to antiviral response through viperin by disturbing viral entry and egress. Reduced Ch25H expression in PGN-treated astrocytes after infection implies viral invasion of TLR2/2-mediated Ch25H expression.

TLR2-induced IFN response was determined by the expression of ISGs that influence cellular membrane properties, which act as an antiviral mechanism. Neuronal and astrocyte cells were treated with TLR2 ligands and infected with HSV-1F for 8 h and mRNA expression was checked by qRT-PCR. ISG expression in neurons and astrocytes
TLR2 dimers induce ISG expression, OAS2, RNase L, PKR, IFIT1 that target viral RNA
ISGs like PKR, oligoadenylate synthetase (OAS), Rnase L, and IFITs target viral protein synthesis, hence inhibiting its replication (40). Upon viral infection, dsRNA is produced during its life cycle and activates 2′-5′-OAS that synthesizes oligoadenylates, which activate latent monomeric Rnase L into potent dimeric ribonuclease. Rnase L cleaves messenger RNA (mRNA) and ribosomal RNA (rRNA), inhibiting the translation and viral replication (21,44). It has been reported that RNase L selectively cleaves viral RNA, but cellular mRNA is also cleaved by the RNase L. Another way to eliminate the virus is to terminate the infected cell; RNase L can induce apoptosis in virus-infected cells (9). As HSV infects and establishes latent infection in the neuron, eliminating neuron cannot be an option. In the trigeminal ganglia culture system, RNase L was found to be protective against HSV-1 (1,8). OAS2 expression was reduced after stimulation with FSL-1, but increased after infection in PGN-stimulated cells; RNase L expression was reduced after stimulation with TLR2 ligands, but expression remained unchanged in HSV-1-infected neurons (Fig. 5A, B). These observations suggest that OAS2 is induced by TLR2/2 stimulation in neurons and assists in the activation of RNase L that cleaves viral RNA. Expression of OAS2, OAS3, and RNase L was suppressed in astrocytes after infection (Fig. 5C–E). Upon stimulation with TLR2 ligands, the expression of these genes was increased to a level higher than that observed for infection alone, but less than uninfected control. In TLR2/2-stimulated astrocytes, OAS2, OAS3, and RNase L were upregulated, but these were suppressed after HSV-1 infection, suggesting that TLR2-TLR2 activation leads to activation of RNase L-mediated viral RNA degradation that can be inhibited by HSV-1.
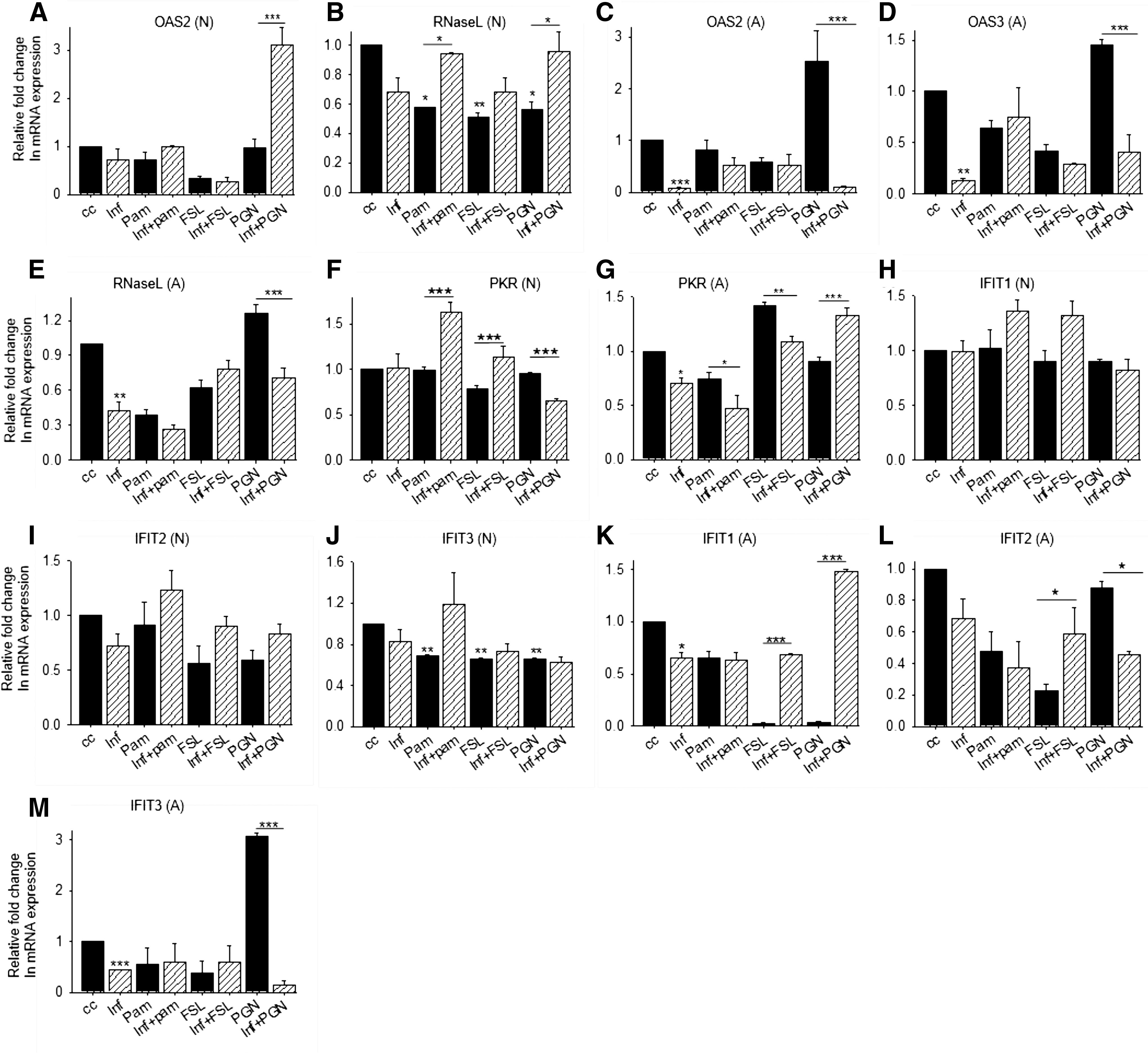
TLR2-induced IFN response was determined by the expression of ISGs that facilitate viral RNA degradation or inhibit viral protein synthesis. Neuronal and astrocyte cells were treated with TLR2 ligands and infected with HSV-1F for 8 h and mRNA expression was checked by qRT-PCR. ISG expression in neurons and astrocytes
PKR is induced by IFNs and activated by recognizing 5′ triphosphate containing viral RNA with the limited double-strand region and dsRNA. When autophosphorylated, PKR inhibits eukaryotic initiation factor (eIF) 2α, and thereby translation. PKR overexpression leads to inhibition of global translation, but ISGs containing 5′untranslated region (UTR) escape from this inhibition (14,36). PKR activity is blocked by HSV proteins such as US11 by preventing its activation by dsRNA binding, while HSV-γ34.5 recruits cellular phosphatases that dephosphorylate eIF2α and rescue the translational inhibition (17,27). In neurons, PKR expression remained unaltered after TLR2/2 ligand treatment, but its expression was upregulated in TLR1/2-stimulated neurons, while it reduced in TLR2/2-stimulated, HSV-1 infected neurons (Fig. 5F). In astrocytes, PKR expression was reduced after TLR1/2 stimulation and was unaltered after TLR2/2 and TLR2/6 stimulation, but increased slightly in TLR2/2-stimulated astrocytes after infection compared to infection alone (Fig. 5G). TLR2/2 stimulation may thus rescue the suppressed PKR expression after HSV-1F infection.
IFIT proteins recognize 2-O methylation of first ribose, 5′ triphosphate, AU-rich sequences and internal ribosome entry site (IRES) elements of viral mRNAs, which are required for cap-independent translation. IFIT1 recognizes viral mRNA and inhibits the preinitiation complex formation either by binding to eIF3c or by blocking the eIF4E from cap binding and hence preventing the viral mRNA translation (11,24,30,35). IFIT1 expression was increased in HSV-1-infected neurons after TLR1/2 and TLR2/6 stimulation, while IFIT2 and IFIT3 expression were reduced after infection and stimulation with TLR2 ligands (Fig. 5H–J). IFIT1 expression was increased in TLR2/2-stimulated astrocytes after infection, while IFIT3 was upregulated upon TLR2/2 stimulation, but gets suppressed after HSV-1 infection (Fig. 5K–M). These observations suggest that IFIT1 is an important ISG during HSV-1 innate response as it can stall viral protein synthesis and is induced by different TLR2 dimers in neurons and astrocytes.
ISG-15 expression repressed in HSV-1-infected astrocytes
ISG-15 is a ubiquitin-like modifier. It is activated by UBE1L forming a thioester bond with ISG15 and transferred to active site cysteine of conjugating enzyme E2L6; finally, E3 ubiquitin ligase HERC6 facilitates the conjugation of ISG15 with target protein (32,47,50). ISG expression was unchanged in neurons after infection or TLR2 stimulation (Fig. 6A), but it was reduced after HSV-1 infection in TLR1/2- and TLR2/6-stimulated astrocytes (Fig. 6B), indicating that ISG15-mediated viral protein degradation is intervened by HSV-1.

TLR2-induced IFN response was determined by the expression of ISGs that facilitate viral protein degradation. Neuronal and astrocyte cells were treated with TLR2 ligands and infected with HSV-1F for 8 h and mRNA expression was checked by qRT-PCR. ISG expression in neurons and astrocytes
TLR2/2 induces Mx-1 expression in HSV-1-infected astrocytes
Mx-1 binds important viral components, directs them to the alternative sites in the cytoplasm. and thereby blocks their function (16,49). Mx-1 expression was unaltered in neurons. while significantly increased after infection in PGN-treated astrocytes. This observation suggests that Mx-1-mediated HSV-1 protein sorting may lead to their degradation and hinder HSV-1 assembly and virion formation that result in reduced HSV-1 replication (Fig. 6C, D).
TLR2/2 suppresses IL-10 expression in neurons and astrocytes after HSV-1F infection
HSV-1 interferes with the cellular machinery to facilitate its replication and suppresses immune response (17,27). Immune suppression is characterized by the elevated levels of anti-inflammatory cytokine IL-10 (13,46). We checked the expression of anti-inflammatory cytokine IL-10 in TLR2 ligand-treated and HSV-1-infected cells. We observed that IL-10 expression levels were not changed after infection in neurons and astrocytes. Upon TLR1/2 ligand treatment, IL-10 expression level was reduced in neurons, while it was undetectable after TLR2/6 and TLR2/2 ligand treatment stimulation. These observations implicate TLR2 in proinflammatory response in neurons. In HSV-1-infected neuronal cells, IL-10 expression was increased in TLR1/2 ligand- and TLR2/6 ligand-treated cells, while it was suppressed in TLR2/2 ligand-treated cells after infection (Fig. 7A). TLR2/2-mediated signaling may therefore help to maintain proinflammatory state after infection in neurons. In astrocytes, IL-10 expression was reduced in TLR2/6- and TLR2/2-targeted cells (Fig. 7B). These observations suggest that TLR2/2 signaling induces proinflammatory state in astrocytes and maintains the same after HSV-1F infection.

Anti-inflammatory cytokine IL-10 expression in HSV-1-infected neurons and astrocytes. Neuronal and astrocyte cells were treated with TLR2 ligands and infected with HSV-1F for 8 h and IL-10 mRNA expression was checked by qRT-PCR. Plotted as relative fold change in mRNA expression (Y-axis) in given treatment/infection (X-axis) compared to untreated control. IL-10 expression in neurons
Discussion
TLR2 recognizes HSV-1 through viral glycoproteins (23). Upon interaction with glycoproteins, TLR2 is activated and subsequent signaling can take place through TLR1/2, TLR2/2, or TLR2/6 dimers inducing cytokines, IFNs, and ISGs that subsequently establish proinflammatory state, terminating HSV-1 infection. In this study, we have observed that TLR2/2 signaling predominantly induced IFN-β in neurons, while it induced IFN-α, IFN-κ, and IFN-ɛ in astrocytes after HSV-1F infection. Moreover, TLR2/2 ligand suppressed expression of IL-10 after HSV-1 infection, whereas TLR1/2 ligand heightened its expression in neurons, while unaffected in astrocytes. The IFNs were mainly induced by TLR2/2 ligand after infection; these IFNs then induce the expression of ISGs like viperin, PKR, OAS2, and IFIT1 in neurons, while they induce viperin, Ch25H, PKR, IFIT3, and Mx1 in astrocytes; these proteins have direct or indirect antiviral functions targeting HSV-1 entry and protein synthesis and egress.
The TLR2-induced innate immune response during HSV infection has been reported by earlier studies stating its protective as well as detrimental role. TLR2 has been shown to induce proinflammatory cytokines and chemokines in HSV-1-infected microglial cells (3). Stimulation of TLR2 on T-reg cells reversed its regulatory function and thus promoted the protective function of HSV-1-specific effector CD4+CD25+ T cells (33). Microglial cells in TLR2 knockout mice had reduced apoptosis and cell deaths 24 h after HSV-1 infection and had downregulated expression of proapoptotic genes (2). TLR2 contributed to lethal encephalitis during HSV-1 infection by enhancing MCP-1 expression and inflammation in the brain and related the mortality to TLR2 activation and not to the HSV-1 levels in mouse brain (22). These findings were supported by the observations that TLR2 on microglia induces an inflammatory response, ROS, and oxidative stress leading to neuronal damage after HSV-1 infection (18,52).
Inflammation in CNS can lead to encephalitis and neuronal damage due to immune hyperactivation and apoptosis (22). Myd88-deficient mice succumb to HSV-1 infection due to uncontrolled viral titer in CNS and TLR2-mediated innate immune response is necessary to control HSV-1 infection (28,43). T cells and NK cells are also involved in protection against HSV-1 (20). So the balanced immune response is the key to combat HSV-1 in CNS. Earlier studies have focused on microglial response during HSV-1 infection, particularly, their potential to establish a proinflammatory antiviral state by secreting cytokines and chemokines as those are the major immune cells in CNS (3,18). HSV-1 establishes latent infection in neurons and can infect astrocytes (10,25). Innate immune response by these cells upon HSV-1 infection that includes induction of cytokines and IFNs subsequently induces the expression of ISGs having antiviral functions and may lead to containment of the virus within these cells or its clearance. We examined whether differential TLR2 response was due to the signaling through different dimers. We observed that major IFN and ISG responses were induced through TLR2/2 dimer upon infection that critically suppressed IL-10. If TLR2-induced proinflammatory response exaggerates the outcome of HSV-1 infection in mice, the tuning of TLR2 response during the infection is necessary. Immunomodulation at a critical time after infection, but before excessive proinflammatory response by using antagonists, preferably a TLR2/2-ligand, as it suppresses IL-10 after stimulation and maintains a proinflammatory state after infection that can be detrimental if persisted longer. The immunomodulation of innate response has been proved helpful in mice where TLR9 antagonist was given on third day after HSV-1 infection, which increased the survival of mice likely by suppressing detrimental proinflammatory immune response (7). On the contrary, the major innate players like IFN and ISGs were induced upon TLR2/2 stimulation in HSV-1-infected cells, proving its importance among TLR1/2, TLR2/2, and TLR2/6 dimers during infection. Considering the potential of TLR2/2 ligand inducing innate response by means of IFNs and ISGs and proinflammatory state by repressing IL-10, the timely induction and repression of TLR2/2 response after HSV-1 infection may lead to favorable outcome.
Footnotes
Acknowledgment
We thank Dr. Jayasri Das-Sarma, IISER Kolkata, India, for her gift of astrocyte cell line DBT.
Author Disclosure Statement
No financial interests exist.
Funding Information
Y.D.B. was awarded a fellowship from the Department of Biotechnology, India. B.S. was awarded a fellowship from the Department of Science and Technology, India (JCB/2018/0027). D.C. received a grant from the Department of Science and Technology, India (DST-SERB, EMR/2015/002118).