
Abstract
Intense immunological dysregulation including immune cell lesions has been characteristically observed in severe cases of coronavirus disease-2019 (COVID-19), for which molecular mechanisms are not properly understood. A study of physiological expressions of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) host cell entry-related factors in immune system components may help explain molecular mechanisms involved in COVID-19 immunopathology. We analyzed transcriptomic and proteomic expression metadata for SARS-CoV-2 host cell entry receptor ACE2 and entry associated proteases (TMPRSS2, CTSL, and FURIN) in silico across immune system components including the blood lineage cells. ACE2 was not detected in any of the studied immune cell components; however, varying transcriptomic and proteomic expressions were observed for TMPRSS2, CTSL, and FURIN. Nondetectable expressions of SARS-CoV-2 host cell entry receptor ACE2 in immune system components or blood lineage cells indicate it does not mediate immune cell lesions in COVID-19. Alternative mechanisms need to be explored for COVID-19 immunopathogenesis.
Introduction
The ongoing pandemic of coronavirus disease-2019 (COVID-19) caused by the novel strain of coronavirus, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has affected millions of lives globally. SARS-CoV-2, an enveloped positive sense single-stranded RNA virus belongs to genus betacoronavirus of the Coronaviridae family that includes SARS-CoV-1 and MERS-CoV: the causative agents of respiratory syndrome outbreaks in recent past (11,14).
Receptor recognition by viruses being the foremost step of viral infections is of paramount importance in determining the severity of infections. In the case of SARS-CoV-2, the viral entry to host cells is reported to be primarily mediated by receptor binding domain of the viral S (spike) protein to a cell surface receptor ACE2. The receptor recognition and the ensuing fusion are critical for viral infections that is facilitated by the entry associated protein transmembrane serine protease 2 (TMPRSS2) or Cathepsin L (CTSL) in concert with FURIN, a host cell associated protease that mediates the cleavage of the viral spike (S) protein (6,7). FURIN has shown to be unique to SARS-CoV-2 and essential for successful viral invasion of the host cells (11).
Epithelial cells enriched in ACE2 expression are selectively biased for bearing the viral load and the COVID-19 infections are primarily attracted to lungs through the nasal route, primarily transmitted among people through respiratory droplets and contact routes. The ongoing studies indicate organ-specific pathogenesis by the SARS-CoV-2 binding-mediated dysregulation of ACE2 (16,22,27). Human ACE2 is an interferon stimulated gene, and thus SARS-CoV-2 may also exploit cell type-specific interferon-driven upregulation of ACE2 (16).
In severe cases of COVID-19 pathogenesis, a hyperactive innate immune response characterized by very high level of proinflammation markers known as “cytokine storm” is observed in lungs as well as other organs. The COVID-19 severity is also characterized by differential response of immune cells: reflected by increased counts of neutrophils and macrophages, however, reduced counts of eosinophil and lymphocytes (specifically of CD4+ and CD8+ T cells) (12,13,19). An increased death of CD4+ and CD8+ T lymphocytes is also a very frequent finding in severe COVID-19 (12,24).
To delineate how the immune system components, especially immune-related blood cells, are affected by SARS-CoV-2 infections of an individual is expected to provide insights into molecular mechanisms involved in immunological dysregulations in COVID-19. Whether the immune system components are potential targets of SARS-CoV-2 or not would depend on the immune system (cell types) expression of the receptor ACE2, and the associated cell entry factors (TMPRSS2 and FURIN).
Currently, studies in humans showing evidence for expression of SARS-CoV-2 putative targets on immune system components are limited, and any evidence at the proteomic level is largely lacking. Recently, Ziegler et al. have performed a collective meta-analysis of human, nonhuman primate, and mouse single-cell RNA-seq data sets for putative SARS-CoV-2 targets (ACE2 and TMPRSS2) (27). The authors noted significant expression of ACE2 in human airway epithelial cells as well as specific cell subsets across tissues, however, not in any immune system components, such as, lymphoid organs, bone marrow, and peripheral blood mononuclear cells (PBMCs). However, their investigation in human tissues was limited to the lung (27). Furthermore, Sungnak et al. analyzed single-cell RNA sequencing data sets from multiple tissues from healthy human donors; however, their investigation into the immune function-related tissues only involved thymus where they found no or little expressions of ACE2 and no coexpression of TMPRSS2 (22). Furthermore, a preliminary study is reported by Muus et al. that presented integrated analysis of available single-cell transcriptomic data sets from human tissues for expression of putative SARS-CoV-2 targets (ACE2, TMPRSS2, and CTSL). The authors noted no significant expression of any of the SARS-CoV-2 host cell entry factors (ACE2, TMPRSS2, and CTSL) in the immune system components; however, any proteomic level analysis was not performed (16). Of note, these studies only analyzed transcriptomic data, and also have not assessed the expression of FURIN in human tissues. Interestingly, in an earlier study by Hamming et al., which employed immunohistochemical analysis, it has been demonstrated that human immune system components lack expression of ACE2 protein; however, authors did not assess expression of any of the other SARS-CoV-2 host cell entry factors, namely TMPRSS2, CTSL, and FURIN (5).
By utilizing the standard databases for transcriptomics (tissue and single cell) as well as proteomic expressions in human immune system components, we have performed a comprehensive in silico analysis for assessing the expression levels of the key SARS-CoV-2 host cell entry receptor ACE2 and cell entry associated host proteases—TMPRSS2, CTSL, and FURIN.
Materials and Methods
We performed in silico analysis of mRNA and protein expressions of ACE2, TMPRSS2, CTSL, and FURIN in human immune system components (bone marrow and lymphoid tissue—thymus, appendix, spleen, lymph nodes, and tonsil) using the tissue transcriptome and immunohistochemistry (IHC) data available in Human Protein Atlas (HPA). IRB approval was not required as we used published metadata for this study. Furthermore, mRNA expression across blood cell types was analyzed using single-cell RNA sequencing data available in “The Blood Atlas (sub-section of HPA).”
External data source methods
Estimation of mRNA expression and localization of human proteins were performed by the source laboratory using tissue and single-cell deep sequencing of RNA (RNA-seq) and IHC in normal tissue.
Immunohistochemistry
As described by the source laboratories, specimens containing normal tissue were collected in accordance with approval from the local ethics committee and also sampled from anonymized paraffin-embedded material of surgical specimens. The specimens were derived from surgically removed tissue: normal was defined by tissue-specific morphological parameters and absence of neoplasia. IHC staining was performed on normal tissue microarray using a standard protocol (10). Antibodies against human ACE2 (HPA000288 and CAB026174), TMPRSS2 (HPA035787), CTSL (CAB000459), and FURIN (CAB009499) were labeled with DAB (3, 3′-diaminobenzidine) stain. Protein expression score was calculated for each receptor based on the staining intensity (negative, weak, moderate, or strong) and fraction of stained cells (<25%, 25–75%, or >75%). For each protein, the IHC staining profile was matched with mRNA expression data and gene/protein characterization data to yield an “annotated protein expression” profile.
Tissue transcriptomics
HPA collects transcriptomic data from the three databases (HPA, GTEx, and FANTOM5). For HPA RNAseq, total RNA was extracted from the tissue samples of healthy individuals (Accession no: PRJEB4337, Ensembl: ENSG00000130234 (version 92.38) using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The extracted RNA samples were analyzed using either an Experion automated electrophoresis system (Bio-Rad Laboratories, Hercules, CA) with the standard sensitivity RNA chip or an Agilent 2100 Bioanalyzer system (Agilent Biotechnologies, Palo Alto) with the RNA 6000 Nano Labchip Kit. Samples of high RNA quality (RNA Integrity Number >7.5) were used for the mRNA sequencing. mRNA sequencing was performed on Illumina HiSeq2000 and 2500 machines (Illumina, San Diego, CA) using the standard Illumina RNA-seq protocol (with a read length of 2 × 100 bases). For estimation of transcript abundance, Kallisto v0.43.1 was used. The normalized tags per million for each gene from the three databases were calculated and included in the HPA. Each tissue was categorized for the intensity of gene expression using a cut-off value of 1 consensus normalized expression (NX) as a limit for detection across all tissues. A tissue was categorized (i) enriched if it had NX level at least four times higher than other tissues, (ii) low specificity if NX ≥1 in at least one tissue, and (iii) not detected if NX <1 in all tissues.
Single-cell transcriptomics
Single-cell transcriptomic data covering various B and T cells, monocytes, granulocytes, and dendritic cells were analyzed. “The Blood Atlas” collects data from the three data sets (HPA (8), Monaco et al. (15), and Schmiedel et al. (21)). HPA presents data for 18 cell types present in human blood that were isolated with cell sorting followed by RNA-seq analysis. Annotation of the studied genes was done according to cell type specificity (cell type enriched, group enriched, cell type enhanced, and low cell type specificity) and cell type distribution (detected in single, few, many, or all cell types).
Further details of the assays and annotation used by the HPA can be accessed online (9).
Metadata analysis
We analyzed HPA transcriptomic and proteomic data in respect of tissue/cell type specific expression in immune system components. Graphs were plotted and inferences were made based on the final results.
Results and Discussion
We have performed in silico analysis of metadata for transcriptomic and proteomic expression of SARS-CoV-2 host cell entry-related factors (ACE2, TMPRSS2, CTSL, and FURIN). SARS-CoV-2 host cell entry receptor ACE2 was not detected in bulk transcriptomic and proteomic expression analysis in any of the studied immune cell components (bone marrow and lymphoid tissue) (Supplementary Table S1 and Fig. 1). ACE2 was also not detected in single-cell transcriptomic analysis of blood cell lineages (Supplementary Table S2 and Fig. 2). “Not detected” to “enriched” transcriptomic expression was observed in bulk (bone marrow and lymphoid tissue) as well as in single-cell analysis (blood cell lineages), and “not detected” to “medium level” proteomic expression (bone marrow and lymphoid tissue) was observed for SARS-CoV-2 host cell entry associated proteases TMPRSS2, CTSL, and FURIN (Supplementary Tables S1 and S2, Figs. 1 and 2).
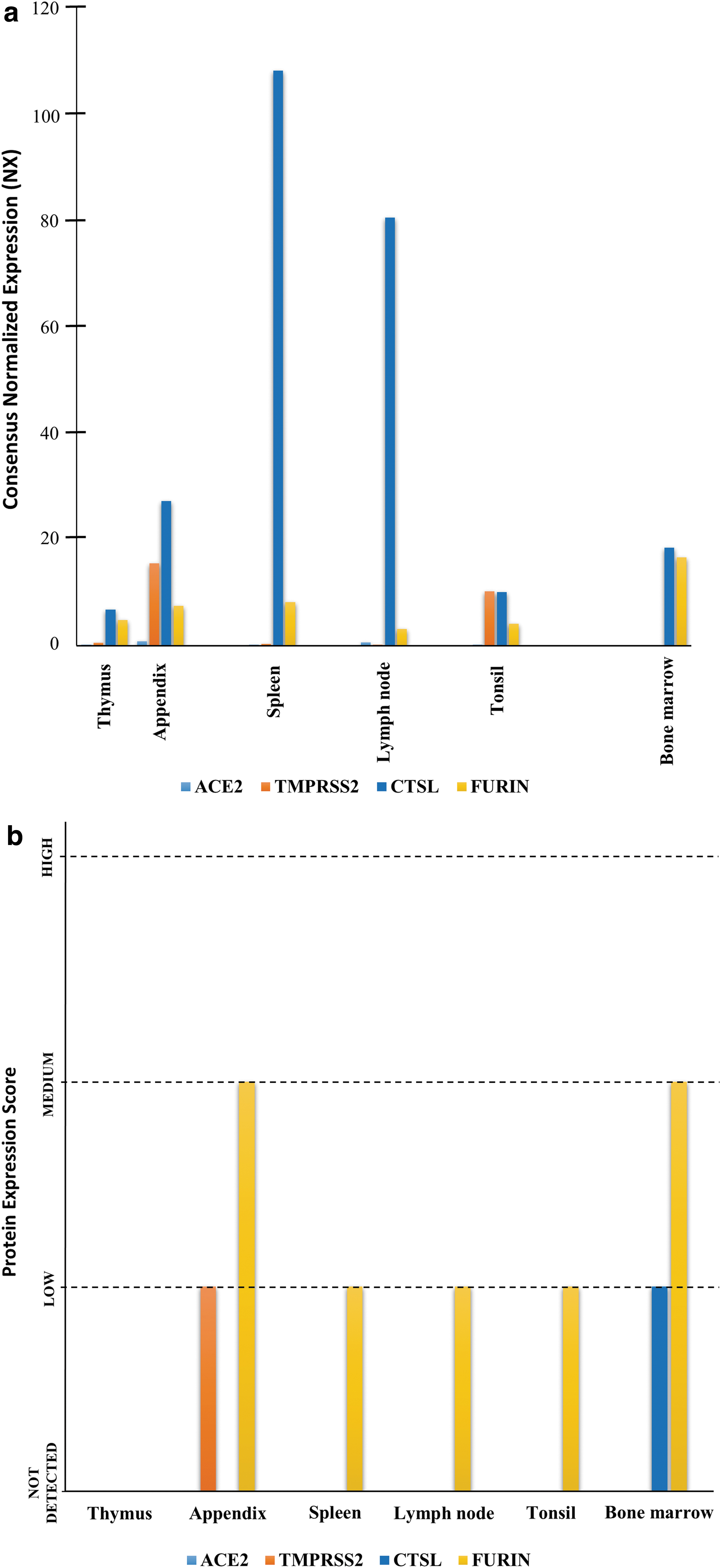
mRNA and protein expressions of SARS-CoV-2 host cell entry factors (ACE2, TMPRSS2, CTSL, and FURIN) in human immune system components (bone marrow and lymphoid tissues).
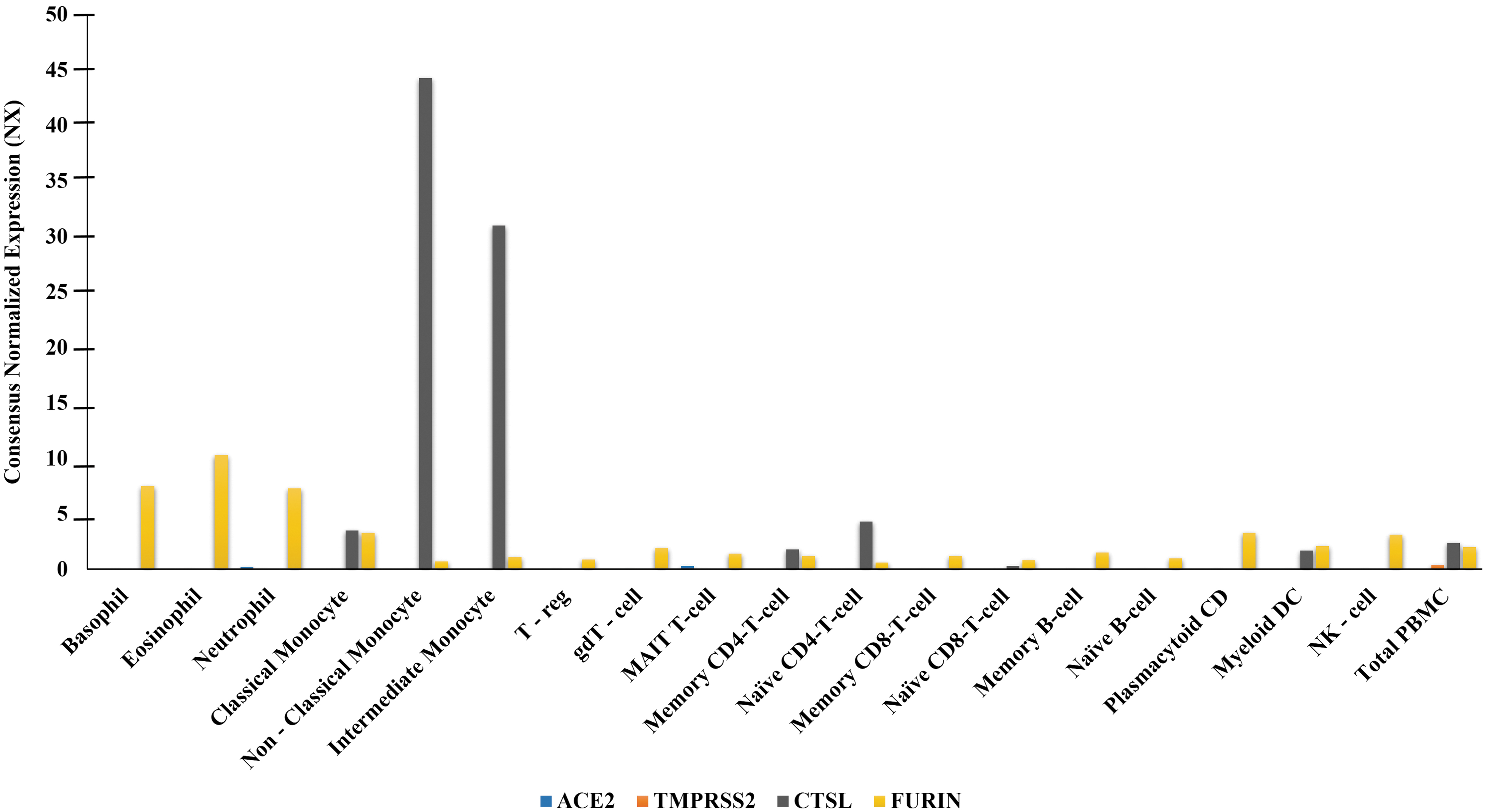
mRNA expression of SARS-CoV-2 host cell entry factors (ACE2, TMPRSS2, CTSL, and FURIN) in human blood cell types. mRNA expression presented as NX. A cell type was categorized (i) enriched if it had NX level at least four times higher than other cell types, (ii) low specificity if NX ≥1 in at least one cell type, (iii) not detected if NX <1 in all cell types. The NX is as described in data source: The Blood Atlas/HPA (8).
The observations made in this study are important and provide insights regarding immunopathogenesis in COVID-19. Our findings of the nondetectable level of expressions of viral host cell entry receptor ACE2 and nondetectable to medium range expression of host cell entry associated proteases TMPRSS2, CTSL, and FURIN across immune system components of healthy individuals (Supplementary Tables S1 and S2, Figs. 1 and 2) are in line with existing studies reporting expression of a single or a set of these factors across human tissues (5,16,22,26). The nondetectable expressions of ACE2 across immune system components including blood (immune) cells indicate their nonsusceptibility for SARS-CoV-2 infection. In exception, a study by Rutkowska-Zapała et al. detected in humans low expression of ACE protein in PBMCs, particularly in a subset of monocytes (CD14++CD16−), but not in other immune cell types (20).
With current level of evidence, host cell expressions of ACE2 and the tissue proteases (TMPRSS2/CTSL and FURIN) are considered essential conditions for SARS-CoV-2 infection (6,7,11), and a high expression of ACE2 alone does not ensure that a cell will be infected (25).
However, this is yet not known whether a low expression of ACE2 is sufficient to mediate viral infection in the presence of the tissue proteases. In addition, no study has yet provided any robust evidence, such as, showing presence of intracellular viral proteins or genetic material, to prove that SARS-CoV-2 can truly infect any of the immune cells. In contrast, recent studies have provided clear indications of immune cell lesion in COVID-19 (3,14,23,26). Lymphocytopenia in COVID-19 cases is marked by selective killing of T cells (26). Moreover, SARS-CoV-2–mediated atrophy and lesion of the human lymphoid tissue—such as spleen and lymph nodes—also have been observed in a recent study (4). The viral-mediated manipulations of the host immune cell receptor signaling are a well-established fact; however, any such evidence specific to SARS-CoV-2 is currently scarce in the literature (18). In absence of significant cell surface expression of ACE2 in immune system components, the alternative mechanisms that are exploited by the COVID-19 virus to induce immunopathology need to be ascertained (12).
Therefore, our finding warrants for a systematic review of existing literature indicated for the set of plausible mechanisms that may explain immunological dysregulation in COVID-19 without the involvement of ACE2 signaling. The available data suggest that selective killing of T cells may be a resultant of either (i) high productions of cytokines and proinflammatory markers (1,3,12), (ii) direct viral protein toxicity (4), (iii) or presence of an additional SARS-CoV-2 cell entry receptor that is expressed in immune system components (2). Possible role of cytokines and proinflammatory markers in killing of T cells received an affirmation from a recent study by Diao et al., which analyzed laboratory data from hospitalized COVID-19 patients (3). The study has reported that T cell numbers were negatively correlated with serum IL-6, IL-10, and TNF-α concentration, and were restored with resolution of the disease, as serum level of these markers was decreased (3). Similarly, possible existence of an alternative host cell entry receptor for SARS-CoV-2 invasion in immune cells received preliminary indications from a recent study by Daly et al., which showed neuropilin-1 (NRP1) mediating SARS-CoV-2 cell entry. The findings show that the FURIN-cleaved S1 fragment of the spike protein binds directly to cell surface NRP1, and blocking this interaction with a small molecule inhibitor or monoclonal antibodies reduced SARS-CoV-2 infection in human cell culture (2). Interestingly, available evidence in human tissues suggests that NRP1 has significant expression in immune cells (17). However, further in situ/in vivo studies will be necessary to validate that NRP1 can mediate SARS-CoV-2 infection of immune cells. In addition, as yet there is no concrete evidence available whether SARS-CoV-2 can infect immune cells; therefore, other mechanisms, such as direct viral protein and cytokine-mediated toxicity (3,4), causing immune cell lesions in COVID-19, particularly of T cells, seem more plausible.
Footnotes
Acknowledgment
Authors express gratitude to “The Human Protein Atlas” for allowing the use of its metadata for this study.
Authors' Contributions
A.K. conceived the idea and wrote the first draft. A.K. and R.K.N. analyzed the data and prepared figures and tables. S.K., R.K.N., C.K., and P.P. reviewed and edited the final draft.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.