
Abstract
Papillomaviruses are placed within the family Papillomaviride, and the members of this family have a double-stranded circular DNA genome. Every year, ∼30% of cancers are reported to be human papillomavirus (HPV) related, which represents 63,000 cancers of all infectious agent-induced cancers. HPV16 and HPV18 are reported to be associated with 70% of cervical cancers. The quest for an effective drug or vaccine candidate still continues. In this study, we aim to design B cell and T cell epitope-based vaccine using the two structural major capsid protein L1 and L2 as well as other three important proteins (E1, E2, and E6) against HPV strain 16 (HPV16). We used a computational pipeline to design a multiepitope subunit vaccine and tested its efficacy using in silico computational modeling approaches. Our analysis revealed that the multiepitope subunit vaccine possesses antigenic properties, and using in silico cloning method revealed proper expression and downstream processing of the vaccine construct. Besides this, we also performed in silico immune simulation to check the immune response upon the injection. Our results strongly suggest that this vaccine candidate should be tested immediately for the immune response against the cervical cancer-causing agent. The safety, efficacy, expression, and immune response profiling makes it the first choice for experimental and in vivo setup.
Introduction
Papillomaviruses are placed within the family Papillomaviride, and the members of this family have a double-stranded circular DNA genome. Proteins E1, E2, E4, E5, E6, E7, E8, E2C, L1, and L2 are involved in viral gene expression, replication, and survival. However, E1, E2, L1, and L2, are found in the entire papillomaviruses identified to date (39) and are essential for processes like replication and shedding of the virus. Papillomaviruses are believed to be host restricted; however, some cases of cross-species transmission are also reported (6,11,12).
The International Human Papillomavirus (HPV) Reference Center has reported >200 HPVs until 2013, and the list is still expanding (5,30). Alpha HPVs are considered as high-risk HPV types (HR HPVs) and are involved in anogenital, head, and neck cancers (4,15). Whereas beta HPVs have a possible role in nonmelanoma skin cancers, in addition to UV radiations (1).
Recent studies have suggested the role of E6 and E7 proteins of mucosal HR HPV types in the increase of cervical cancer by altering the pathways that regulate the human immune system response to promote cellular transformation and cause persistent infection (28). However, its cutaneous expression is also important for cellular transformation initiation and maintenance; meanwhile, for skin cancer, the expression of E6 and E7 is not crucial. Beta HPVs exacerbate the accumulation of somatic mutation and DNA breaks induced by ultraviolet radiations. Hence, it plays a vital role by acting as a facilitator in skin carcinogenesis (11).
Every year ∼30% of cancer cases are caused by HPV (4). HPV16 and HPV18 are reported to be associated with 70% of cervical cancers (41). In this study, we aim to design B cell and T cell epitope-based vaccine using the two structural major capsid protein L1 and L2 as well as other three important proteins (E1, E2, and E6) against HPV strain 16 (HPV16). We used a computational pipeline to design a multiepitope subunit vaccine and tested its efficacy using in silico computational modeling approaches. Our analysis revealed that the multiepitope subunit vaccine possesses antigenic properties and using in silico cloning method revealed proper expression and downstream processing of the vaccine construct. Besides this, we also performed in silico immune simulation to check the immune response upon the injection. Our results strongly suggest that this vaccine candidate needs further scientific consensus by testing in experimental setup to be used against the cervical agents.
Materials and Methods
Retrieval of HPV16 polyprotein
Using the GenBank database of the NCBI (
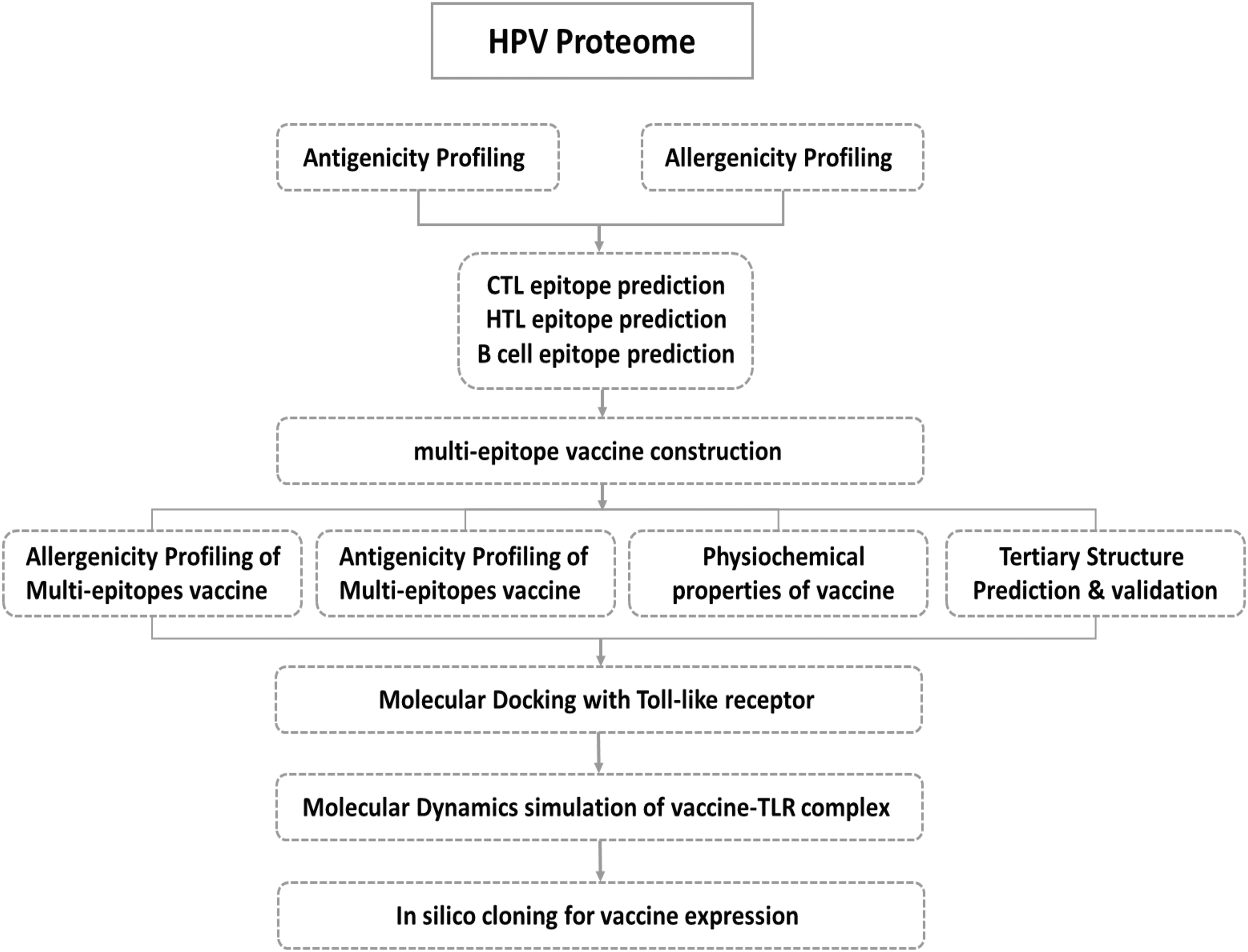
Systematic flow diagram of the vaccine design for HPV.
Cytotoxic T lymphocyte epitope prediction
NetCTL 1.2 (
Helper T lymphocyte epitope prediction
The Immune Epitope Database (IEDB) tool (
Toxicity prediction
The predicted epitopes were submitted to ToxinPred (
Construction of multiepitope vaccine sequence
The final predicted CTL and HTL epitopes, filtered through the discussed immunoinformatics strategy was used to construct a sequence. Beta-defensin was used as an adjuvant (16), and the epitopes were joined using AAY linkers for CTL epitopes and GPGPG linkers for HTL epitopes, respectively (34).
Allergenicity prediction of the vaccine
AlgPred (
Antigenicity prediction of the vaccine
VexiJen (
Physiochemical properties prediction
The physiochemical properties for the constructed sequence were analyzed with the help of the ProtParam web tool (
Secondary structure prediction for the final vaccine sequence
PSIPRED tool (
Tertiary structure prediction
Robetta (
Tertiary structure refinement
Galaxy Refine tools (
Tertiary structure validation
ProSA-web (
B cell epitope prediction
BCPred (
Molecular docking of vaccine with the receptor
HDOCK (
In silico cloning and optimization of vaccine protein
In the JCat tool (Java codon adaptation tool) (13) was used to express the B cell and T cell epitope-based vaccine in a suitable vector for codon optimization, expression, and reverse translation. The codon optimization was significant for the expression of vaccine structure in a host Escherichia coli (strain K12) as the usage of a codon is comparatively different in E. coli than the native host. Rho-independent transcription, restriction cleavage sites, and prokaryotic ribosome-binding sites were eluded through considering three extra options. Java codon adaptation tools provide the output in terms of CAI (codon adaptation index) and %GC content to confirm the high level of protein expression. For cloning, the final vaccine in E. coli pET-28a (+) vector modification N and C terminal with XhoI and NdeI restriction sites was performed, respectively. Finally, for expression, the prepared optimized sequence, along with the restriction sites, was installed to the pET-28a (+) vector utilizing the SnapGene tool.
Immune simulation
To understand the dynamics of the human immune system in response to foreign particles, a server that uses agent-based modeling, C-ImmSim, was used to predict the relationships between the human immune system and the foreign particle (33). Production of cytokines as well as other substances like interferon and antibodies were estimated by applying the Position-Specific Scoring Matrix method. Moreover, response for T helper cell 1 and T helper cell 2 (Th1 and Th2) was also predicted with the defaulted parameters measure of diversity or Simps Index by the server.
Vaccine population coverage
To crosscheck the impact of amino acids substitution on vaccine population coverage. BLAST was performed, and the sequences were retrieved for alignment. ClustalW was used to perform sequence alignment. The predicted epitopes were then mapped and checked for any mutations. In our search, we selected global sequences to make sure that the vaccine is universal.
Results
Protein collection
In this study, the most suitable potential candidate proteins of HPV strain 16 (HPV16) were retrieved from UniProt under the UniProt ID: UP000009251 for designing B and T cell multiepitope vaccine using immunoinformatics approach. HPV16 proteome comprises a total of nine proteins out of which five proteins, Replication protein E1, Protein E6, Minor capsid protein L2, Regulatory protein E2, and Major capsid protein L1, were used after antigenicity analysis as shown in Table 1.
List of Candidate Proteins with Antigenicity Score Predicted Using VexiJen Server
Prediction of CTL epitopes
For all the 5 proteins, a total of 73 CTL epitopes were predicted. Out of which 18 CTL epitopes for Replication protein E1, 4 for Protein E6, 19 for Minor capsid protein L2, 13 for Regulatory protein E2, and 19 CTL epitopes for Major capsid protein L1 were predicted. However, based on toxicity, allergenicity, immunogenicity, and MHCI binding affinity, only three epitopes were selected (Table 2).
List of Final Nontoxic and Nonallergen Cytotoxic T Lymphocyte Epitopes with Their Combined and Immunogenicity Score, Respectively
CTL, cytotoxic T lymphocyte.
Prediction of HTL epitopes
HTL epitopes were finalized the same way as CTL epitopes, as shown in Table 3, and used to construct the multiepitope vaccine. For HTL epitope prediction, seven reference set of Human alleles was used. The final HTL epitopes for the five proteins positioned as follows:450–464 (HLA-DRB5*01:01), 448–462 (HLA-DRB5*01:01), 128–142 (HLA-DRB5*01:01), 126–140 (HLA-DRB5*01:01), 281–295 (HLA-DRB5*01:01), 459–473 (HLA-DRB3*02:02), 255–269 (HLA-DRB1*03:01) 175–189 (HLA-DRB3*02:02) 112–126 (HLA-DRB3*01:01), and 422–436 (HLA-DRB3*01:01).
List of Selected Helper T-Lymphocyte Epitopes with Its Respective Percentile Rank Predicted Using The Immune Epitope Database Tool
B cell epitope prediction
A total of 10 linear B cell 20mer epitopes were predicted for the constructed vaccine using the BCpred server with the defaulted parameters (Table 4). Conformational B cell epitopes with a > 0.5 score were considered discontinuous (Table 5). The 3D model of the vaccine showing linear and discontinuous B cell epitopes is shown in Figure 2A. The ElliPro score represents the average value of PI for the epitope residues. The acquired probability score also approves and confirms the immunological behavior of the designed multiepitope subunit vaccine. ElliPro-predicted epitopes as shown on the vaccine 3D model is given as Figure 2B.
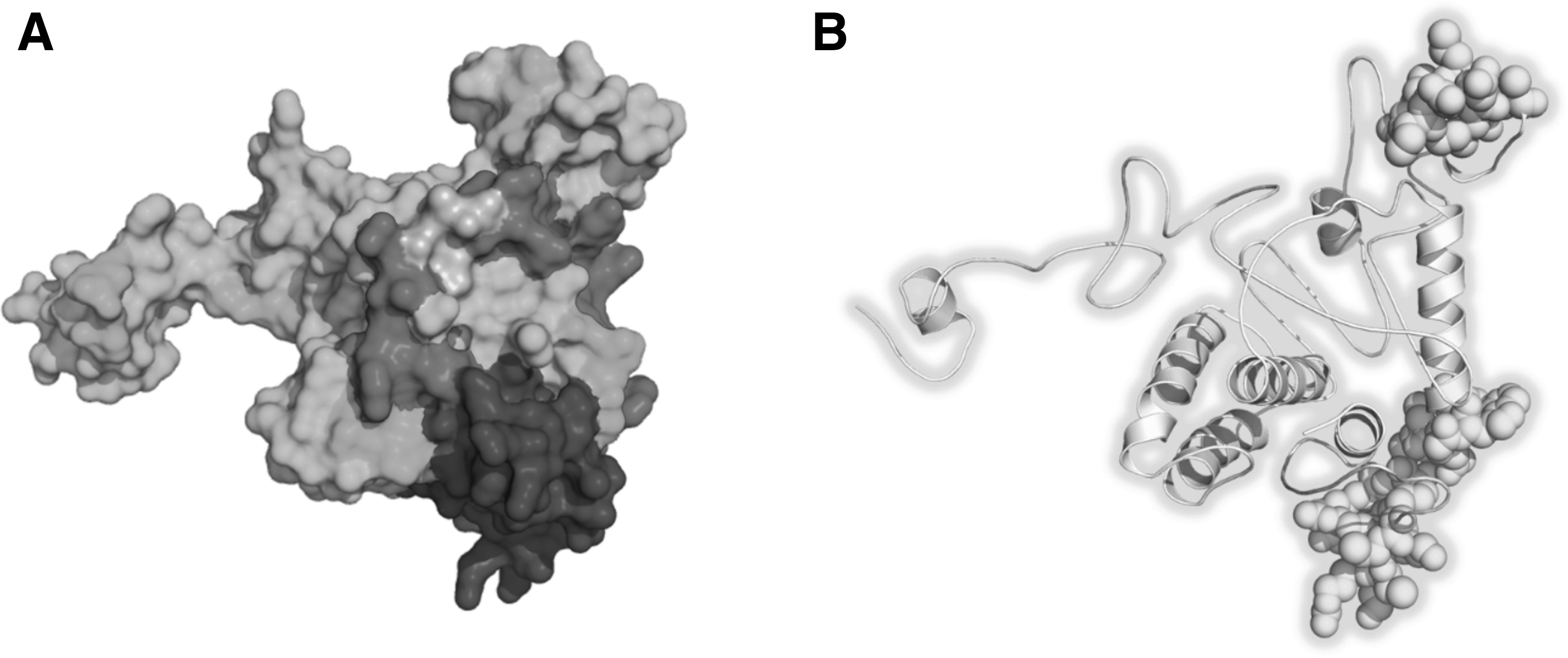
B Cell Epitopes Predicted by BCpred with Defaulted Parameters
B-Epitope Residues Predicted through Ellipro
Designing a final vaccine construct
Five CTL and 10 HTL epitopes were used to construct a subunit vaccine sequence linked with a ratio of 1:2. Adjuvant was linked to the first CTL epitope N-terminal with EAAAK linker, and the rest of the CTL epitopes were joined using AAY linkers. The last CTL epitopes were fused with the HTL epitope using a GPGPG linker. At last, all the HTL epitopes were fused using GPGPG linkers. Schematic presentation of the complete vaccine unit comprising a multiepitope peptide and linked adjuvant is shown in Figure 3.
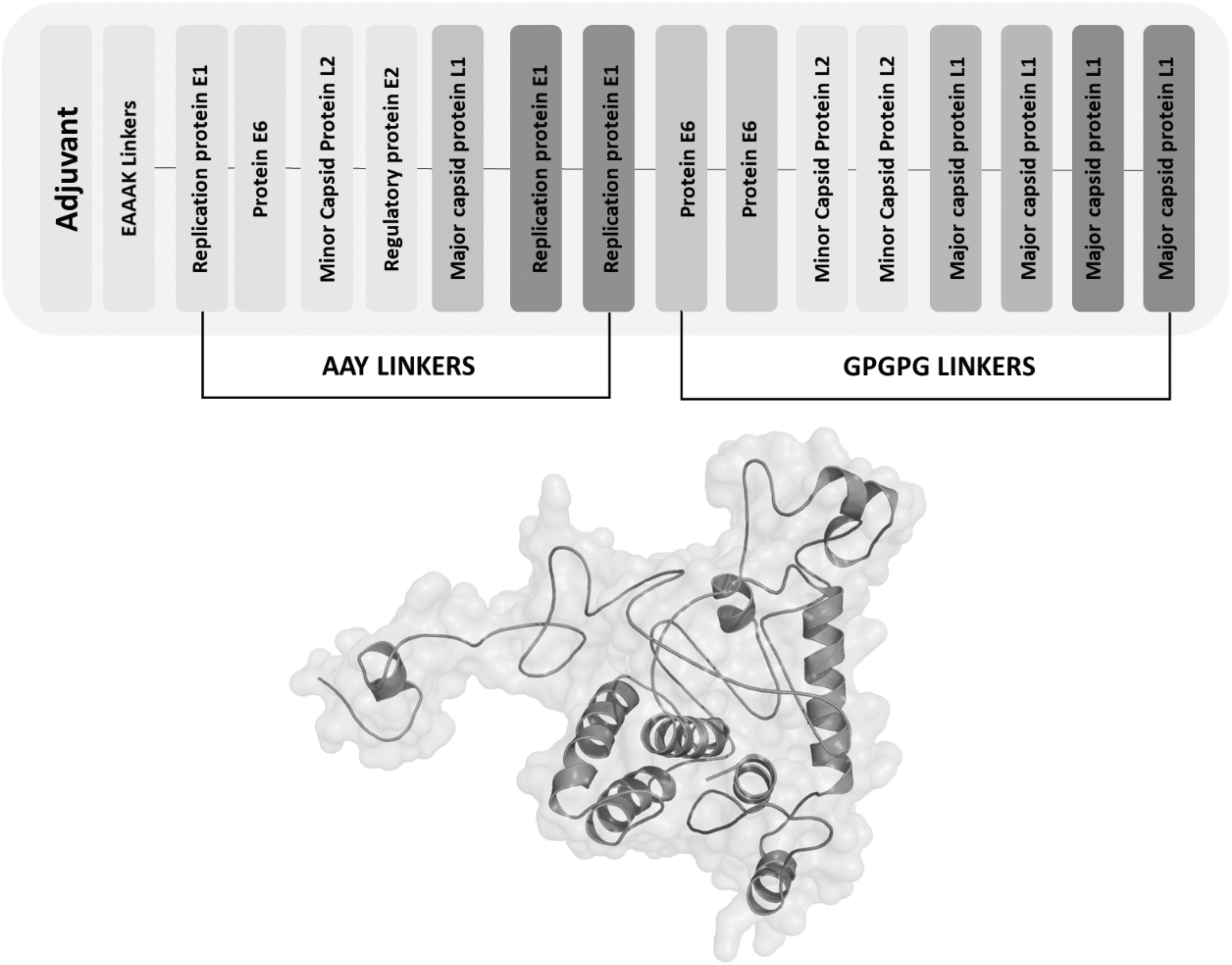
Schematic presentation of the complete vaccine unit comprising a multiepitope peptide and linked adjuvant. Epitopes of a single protein are shown by the same color, whereas CTL and HTL epitopes are linked by separate linkers. The vaccine 3D structure (brown cartoon ribbon) given is also illustrated.
Three-dimensional structure prediction, validation, and refinement
The 3D structure for the constructed sequence was developed using the Robetta server (36). The server-initiated five models, out of which the best was selected was based on poststructure evaluation parameters. The five refined models provided by Galaxy Refinement server, the best model (model 1) was selected based on various factors like calculated RMSD, Mol Probity, High GDT-HA score Poor rotamers, and Rama favored. Out of five refined models provided by the server, model 1 was selected as a refined structure and used further in docking. Model 1 3D structure is provided in Figure 3. It was decided based on numerous factors, such as a high GDT-HA score of 0.9829, RMSD calculated was 0.317. The Mol Probity was measured by 1.571. The clash score measured atoms' overlapping, which was low for model 1 and was calculated as 11.0.
Similarly, Poor rotamers were calculated as 0.0, and the Rama favored was calculated as 99.3, which were far better than other models, as shown in Table 6. Therefore, the refined model 1 was selected for further analysis. The selected 3D model of the vaccine was then subjected to a structure assessment phase. ProCheck for protein model showed that 95.2% of amino acids are plotted in the most favorable areas, 4.8% in allowed regions, and only 0.0% in disallowed regions. Similarly, ERRAT and ProSA-web calculated and verified the overall quality of the basic 3D model. The resulting overall quality factor reported by ERRAT was 79.7%, and the ProSA-web showed a score of −4.58 for the 3D model of vaccine construct, which proved the accuracy of the model. Results from ProSA-web and rampage are given in Figure 4.
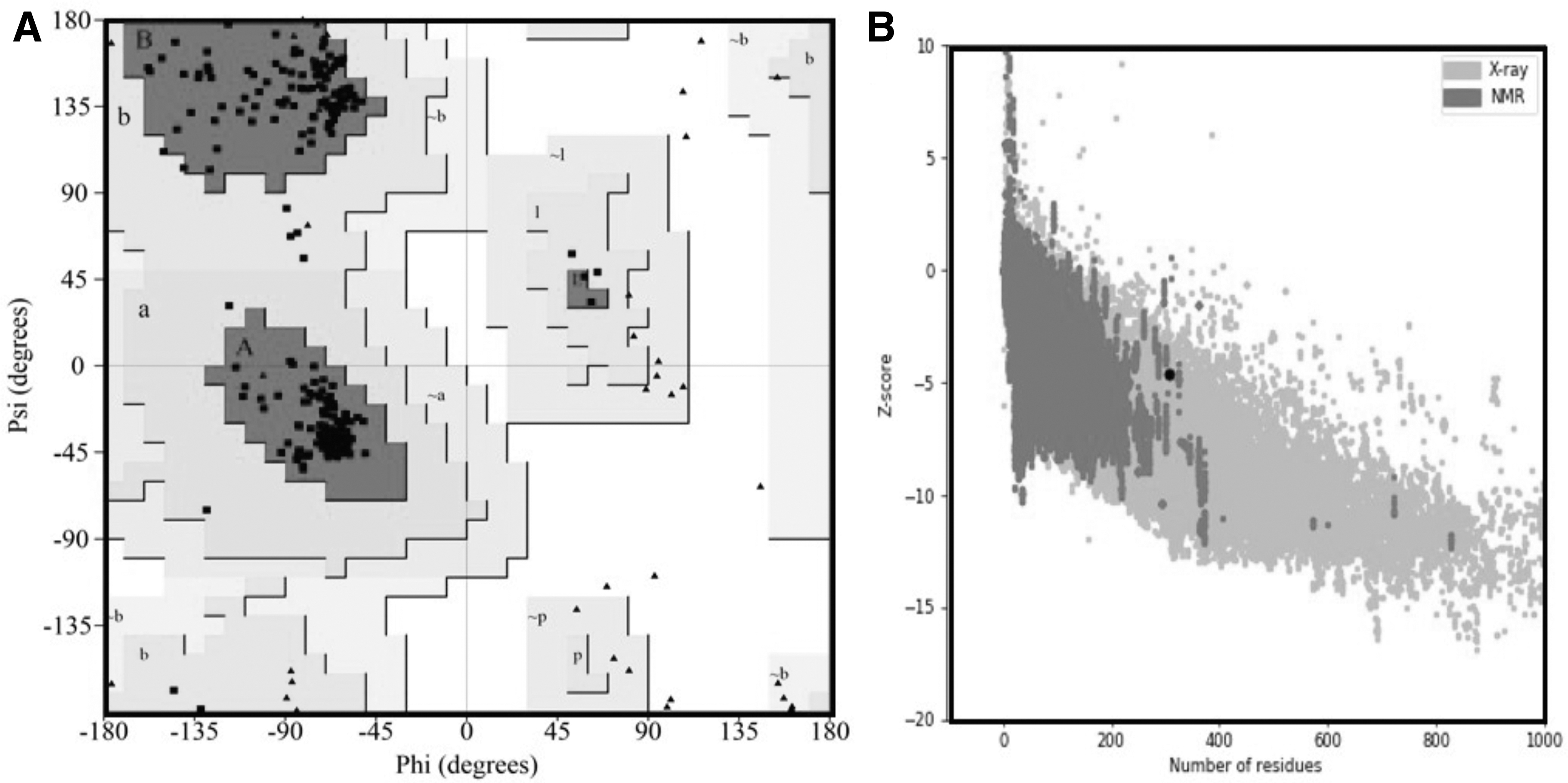
Quality assessment of the vaccine model using rampage Ramachandran plot
Galaxy Refined Results for Vaccine Construct
The model was selected for its low MolProbity and high Rama-favored score.
Antigenicity and allergenicity of vaccine
Antigenicity and Allergenicity of the vaccine sequence were predicted using the VexiJen server and AllerTop server. VexiJen classified the sequence as Antigenic with a score of 0.4871 and AllerTop as nonallergen with the nearest protein in UniProtKB accession number Q86UU0 defined itself as nonallergen.
Physiochemical properties of vaccines
ProtParam predicted physicochemical properties of the proposed vaccine sequence as molecular weight 33.72 kDa, theoretical PI 9.58, which classify it as basic in nature. In vitro half-life in mammalian reticulocytes was estimated as 30 h: in vivo in yeast as >20 h and in vitro in E. coli as >10 h. The predicted instability index was 39.50, which represents the stability of the vaccine. The aliphatic index of the vaccine sequence was computed to be 59.80, indicating thermostability, and a negative GRAVY −0.521 represents the hydrophilic nature of the constructed sequence.
Prediction of secondary structure
The secondary structure of the vaccine construct can be categorized as; alpha-helix (Hh) 34.4%, extended strand (Ee) 21.06%, and random coil (Cc) 55%. Graphically, secondary structure elements are provided in Figure 5.

Graphical representation of the secondary structure of the vaccine construct.
Molecular docking of constructed vaccine with human toll-like receptor 3
From a previous literature review showing the involvement of Human TLR3 in HPV-related diseases and infections, the docking of the vaccine was performed with TLR3 (PDB ID: 1ziw). Docking of both the molecules was performed through ClusPro 2.0. From the 30 models created, only 1 model on the basis of low energy and large size was selected as it showed better interaction between the receptor and ligand. Docked complex intermolecular conformation and chemical interactions are presented in Figure 6.
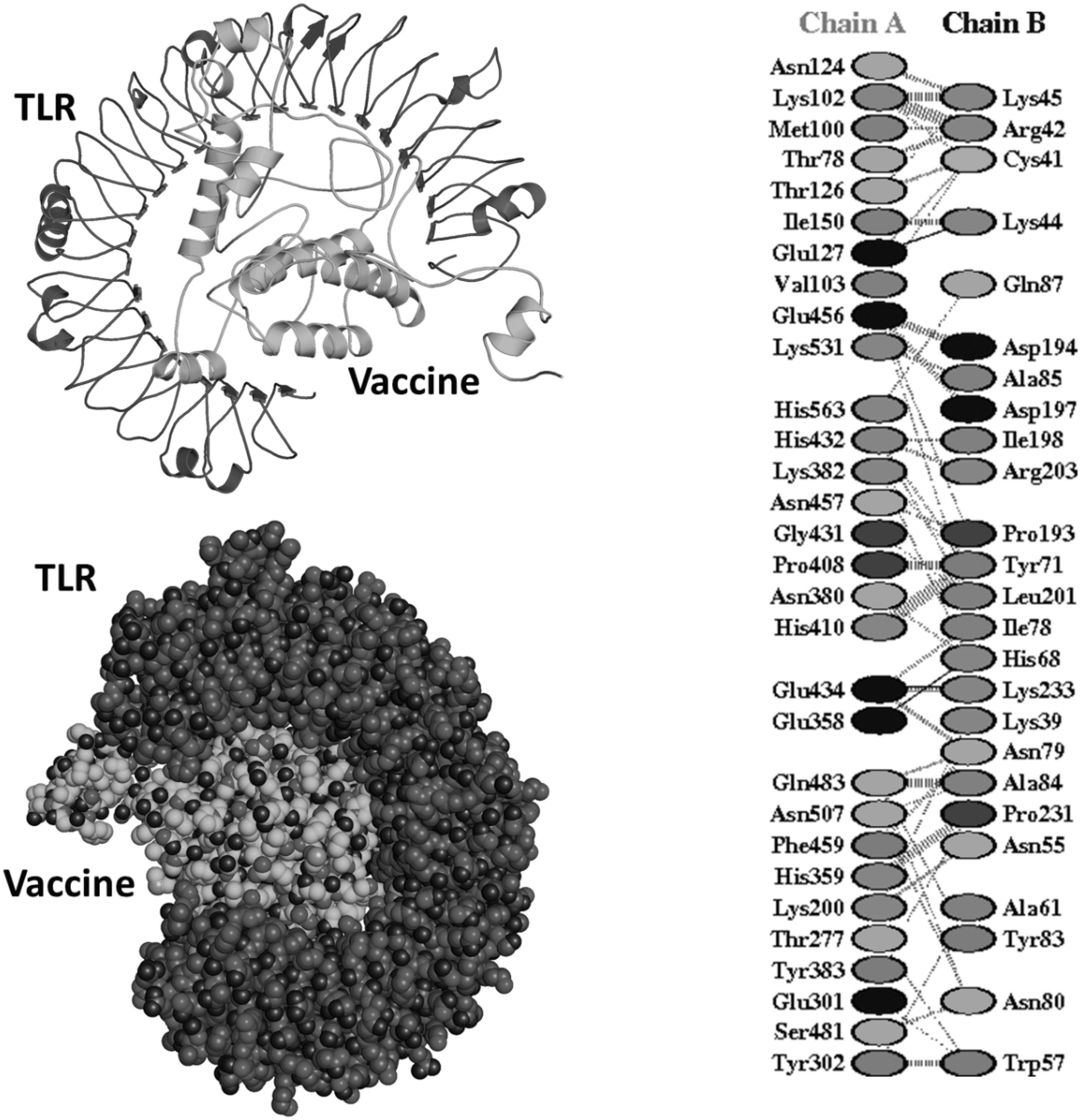
Top-ranked model of docking assay. The different types of chemical interactions involving residues from TLR3 receptor and vaccine are also provided.
Codon adaptation and in silico cloning
The amino acid sequence of the vaccine was uploaded to JCAT to adapt vaccine sequence codon usage according to E. coli K12 strain. The calculated CAI was 0.96, with the GC contents 56.24%. Such results indicated a better expression level of vaccine construct in E. coli (K12 strain). GC contents between 35% and 70% have been reported for better expression of the gene. Finally, the optimized vaccine was cloned into pET28a (+) plasmid using the restriction cloning tool of SnapGene. For this purpose, suitable restriction enzymes Xh0I and NdeI were selected, and their restriction sites were inserted at both ends of the adapted nucleotide sequence. Restriction sites for Xh0 and NdeI were added at the N-terminal and C-terminal of the optimized vaccine construct and cloned in pET-28a (+) vector (Fig. 7). The total size of the cloned vector is 1,137 bps.

Cloned vaccine sequence (red) into peT-28a vector.
Immune simulation of the proposed vaccine and immune response triggering
The human immune system response was monitored through in silico immune simulation upon the antigen's injection in several doses. The immune response was significantly triggered, and the antibody titer was very high after the injection (Fig. 8A). It can be clearly observed that the combined IgM+IgG titer remained 700,000xx/mL. The IgM alone titer was reported to be around 55000xx/mL, whereas Ig1 was reported as 15,000xx/mL. We also checked the interleukin (IL) and cytokine responses (Fig. 8B). The IFN-g and IL-2 titers were significantly high. This shows the consistent and robust immune triggering response upon the injection. The cellular immune system response to pathogen identification at re-encounter was also robust, including memory cells' development. The maximum concentration of 375 cells/mm3 for the phagocytic natural killer cell population was reported (Fig. 8C). The population of the T cell was reported to be >1,150 cells/mm3 (Fig. 8D). Dendritic cells and phagocytic macrophage populations were 200 cell/mm3 each (Fig. 8E, F). Hence these results confirm that our vaccine candidate effectively triggers the immune response.
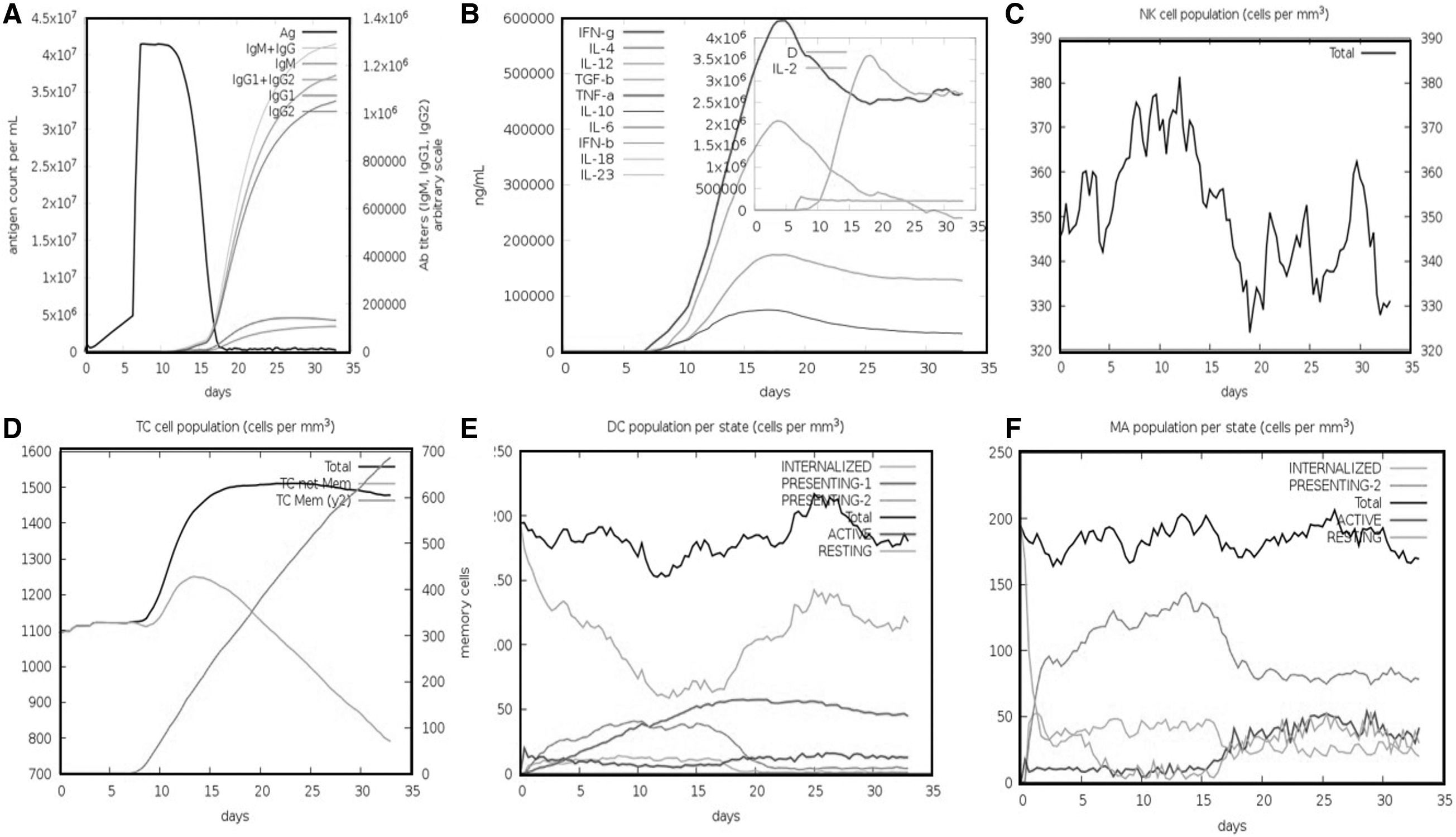
In silico immune simulation by the vaccine followed by injection. The immune response to the vaccine is demonstrated by antibodies titer
Epitopes' conservation analysis
Using BLAST ∼200 different strains from different countries, including United States, India, South Africa, Thailand, United Kingdom, and other countries were analyzed. To check the impact of any mutations reported in different strains, ClustalW-based alignment was performed for each protein. From the alignment it can be seen that all the epitopes in different proteins lie in conserved regions and would vaccine worldwide immunity. All the alignments are shown in Supplementary Figures S1–S5. These analyses suggest that the predicted vaccine has wide population coverage. The sequential tutorial of this methodology is given in the Supplementary File.
Discussion
HPV-related cancers have emerged to be endemic worldwide. They play various types of cancers like head and neck squamous cell carcinomas (HNSCC) penile, anal, and cervical head and neck squamous cell carcinomas. HNSCC have increased the number of incidences in comparison to cervical cancers and is predicted to continue until 2060 (16).
Vaccination plays a substantial role in the effective control of infectious diseases. Epitope-based vaccine designing is beneficial, profitable, highly stable, nontoxic, and easy in engineering (27). Hence, it has the upper hand over conventional vaccines, which may have many apprehensions in immunocompromised individuals (20). These multiepitope-based vaccine peptides are retrieved from the pathogen's proteome and are designed from highly specific and immunogenic B cell and T cell epitopes (2,19,20). Immunization is one of the most effective methods to control infectious diseases. Immunoinformatics approaches are speedy and efficient tools for the identification of potential candidates for vaccine designing from a pathogen's proteome (2,18 –26).
The proteome of HPV strain 16 consists of 9 proteins. This study used L1, L2, E1, E2, and E6 proteins to construct the multiepitope vaccine. Prediction of the best CTL, HTL, as well as B cell epitopes was carried out utilizing different bioinformatics tools. VaxiJen server predicted the antigenicity of vaccine construct to be 0.4871 while its allergenicity was predicted using AllerTop server classifying it as nonallergen with nearest protein UniProt ID Q86UU0 defined as nonallergen. The constructed vaccine's molecular weight was calculated as 33.72 kDa with a theoretical PI of 9.58, showing the basic nature of the vaccine construct. The instability index value was computed to be 39.50 classifying the developed vaccine as stable. The aliphatic index of the subunit vaccine is 59.8, and GRAVY is −0.5, which represents its hydrophilic nature.
Moreover, homology modeling approach was used to generate the tertiary structure of the developed subunit vaccine sequence. Meanwhile, the validation and accuracy check of the tertiary structure was performed with tools like ProSAWeb, ERRAT, and Procheck. Molecular docking of the vaccine and TLR-3 also revealed its strong interaction.
Codon optimization was performed for better expression and efficient transcription and translation of foreign protein in the E. coli K-12 strain. The efficiency of codon optimization was determined by calculating the total GC content and DNA sequence's CAI value. Finally, the vaccine was cloned into pET-28a (+) vector, where XhoI and NdeI restriction sites were inserted at the N- and C-terminals of the adapted codon, respectively. Later on, the optimized codons with restriction sites were introduced into the vector by restriction cloning. To close, a cloned construct was generated, having a sequence a total length of 1,137 bps. Furthermore, immune simulation also provoked the immune response.
Conclusions
The role of HPVs in cancers and other diseases is pronounced. An ultimate protocol is required for coping with the situation. In this study, we manipulated the proteome of HPV16 using a different easily available free online server for the prediction of immunogenic CTL, HTL, and B cell epitopes. A vaccine sequence was constructed, modeled, and evaluated for physicochemical properties. Additionally, immune simulation was performed for an insight into the human immune response to the constructed vaccine. Also, the interaction of the proposed construct vaccine with Human-TLR3 was performed, and at last, the sequence was cloned in an appropriate vector for expression. The developed vaccine construct might be able to prevent or lower HPV-related cases.
Footnotes
Authors' Contributions
All the authors were involved in design, analysis, writing and finalizing the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
D.-Q.W. is supported by grants from the Key Research Area Grant 2016YFA0501703 of the Ministry of Science and Technology of China, the National Science Foundation of China (Grant Nos. 32070662, 61832019, 32030063), the Science and Technology Commission of Shanghai Municipality (Grant No.: 19430750600), the Natural Science Foundation of Henan Province (162300410060), as well as SJTU JiRLMDS Joint Research Fund and Joint Research Funds for Medical and Engineering and Scientific Research at Shanghai Jiao Tong University (YG2017ZD14). The computations were partially performed at the Pengcheng Laboratory. and the Center for High-Performance Computing, Shanghai Jiao Tong University.
Supplementary Material
Supplementary File
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.