
Abstract
During viral infections, cells produce type I interferons (IFNs), which are detected by the IFNα/β receptor (IFNAR). To survive in hosts, viruses have strategies to downregulate IFN-mediated signaling. We hypothesized that macrophages, which are among the first myeloid cells to respond to viral infections, would produce a different cytokine profile if responding to ligation of pattern recognition receptors (PRRs) while IFNAR-mediated signaling was compromised. Specifically, IFNAR-mediated regulation of interleukin (IL)-1α, IL-6, IL-12, and tumor necrosis factor-α was studied in cultured murine bone marrow-derived macrophages. Since viruses like vesicular stomatitis virus can ligate PRRs such as Toll-like receptor (TLR)4 and 7, macrophages were stimulated with the TLR4 and TLR7 agonists lipopolysaccharide (LPS) or imiquimod, respectively, with or without antibody-mediated IFNAR-blockade. Cytokines and viability were assessed for 3 days poststimulation. Blocking IFNARs acutely exacerbated cytokine production by macrophages and aided their survival when they were treated with LPS. In contrast, cytokine concentrations were unaffected or slightly reduced by IFNAR blockade, but macrophages died at a greater rate when imiquimod was the stimulus. This demonstrated a differential role of IFNAR signaling in regulating PRR-induced cytokines. This suggests potential mechanisms whereby macrophages responding to viruses that inhibit type I IFN responses might contribute to excessive inflammation in some cases and inappropriately low-magnitude responses in others. This also provides a well-defined cell-based model for further dissecting the role of type I IFN signaling in macrophages responding to viral and other infections.
Introduction
Macrophages originate as monocytic precursors and are housed throughout the body in locations such as the spleen (17). They get distributed to replenish macrophages in a steady state, as well as during inflammatory conditions. They function to efficiently digest and clear dead and foreign material through phagocytosis and also play a role in the production of inflammatory cytokines (40).
In addition, macrophages are able to mediate inflammation in four stages: recruitment to tissues, differentiation and activation in situ, conversion to suppressive cells, and restoration of tissue homeostasis (49). Macrophages are recognized for their ability to mediate the innate immune response by the phagocytosis of pathogens (14). Signaling through pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) facilitates activation and subsequent maturation of macrophages. Macrophages play roles in a vast array of disease conditions ranging from wound healing to host defense to immunoregulation (40).
Interferons (IFNs) are types of cytokines that play important roles in responses to pathogens and were identified in secretions of virus-infected cells that had a protective effect on neighboring cells (21). IFNs are well recognized for their strong antiviral properties, their ability to regulate cell growth, and their immunomodulatory effects (30). IFNs have long been divided into two distinct groups: type I IFNs and type II IFN, but only recently has a third type, type III, been acknowledged (43). Type I IFNs have both a direct and indirect ability to fight viral infection and are the first line of defense against viruses. Directly, they inhibit viral replication in infected cells, and indirectly, they stimulate adaptive immune responses (33). They are able to regulate the immune system by acting on natural killer cells, B cells, T cells, dendritic cells, or phagocytic cells (36).
TLRs are a membrane-spanning class of PRRs that are key participants in the IFN signaling pathways and regulate the activation of both the innate and adaptive arms of the immune system, both of which are required to respond to microbial pathogens (1). The first mammalian TLR to be identified was TLR4, which recognizes lipopolysaccharide (LPS), a major component of the cell wall of Gram-negative bacteria. TLR4 was also reported to be triggered by infection with retrovirus, respiratory syncytial virus, and mouse mammary tumor virus (28). There is a growing list of viruses that induce an inflammatory response during acute infection through TLR4 activation. Known TLR4-activating viral proteins include the vesicular stomatitis virus glycoprotein (34).
TLR7 recognizes single-stranded RNA (ssRNA) derived from RNA viruses (e.g., vesicular stomatitis virus, influenza viruses, and human immunodeficiency viruses). Because of this ability to recognize ssRNA, TLR7 is also capable of being activated by chemicals that mimic these viruses (e.g., imidazophinoline derivatives like imiquimod and resiquimod and guanine analogs) (37).
Key players in mounting an appropriate immune response are antigen-presenting cells. This group of cells includes both macrophages and dendritic cells, which mature upon encountering dangerous antigens and are then able to migrate from the periphery to T cell-rich secondary lymphoid tissues (39). Antigen-presenting cells are able to sense pathogen-associated molecule patterns (PAMPs) through ligation of PRRs both on their surface (e.g., for extracellular bacteria) and intracellularly (e.g., for viruses) (5).
As an initial method of host cell defense upon the ligation of PRRs such as TLRs by viral PAMPs, cells are able to initiate a signaling cascade which produces type I IFNs (9). The production of IFNs can be triggered by the ligation of many different receptors. In macrophages and dendritic cells, type I IFN production can be triggered by TLR4, TLR7, TLR8, and TLR9 (2).
These type I IFNs include IFNα and IFNβ and are detected by cells that express the heteromeric IFNα/β receptor (IFNAR). Type I IFN production can be amplified by a positive feedback loop: the early production of IFNβ and IFNα4 induces the transcription of IFN regulatory transcription factor-7 (23), which, once phosphorylated, helps to drive transcription and expression of other genes in the type I IFN family, thus enhancing IFN production (25). IFN has the ability to fight viral infection both directly, by inhibiting viral replication in infected cells, and indirectly, by stimulating the adaptive immune response (6).
Type I IFNs are regarded for their antiviral properties. However, IFNs are not always effective against viruses. To evade these IFN-induced antiviral mechanisms and survive in their hosts, viruses use a plethora of strategies to downregulate the production of type I IFNs and/or the expression of IFNAR and/or downstream signaling components (12,28).
Thus, this study sought to examine the importance of signaling through the IFNAR in the regulation of key cytokines, including interleukin (IL)-1α, IL-6, IL-12, and tumor necrosis factor (TNF)α, all of which play roles in an array of clinical conditions associated with dampened signaling through the IFNAR. IL-1α was studied instead of IL-1β because it has been reported by others that IL-1β tends to upregulate IL-1α, whereas the reverse occurs to a lesser extent (15,24). As such, IL-1α could be considered a downstream effector cytokine in response to IL-1β production. Indeed, IL-1β has been shown to potentiate the secretion of IL-1α (11).
Murine bone marrow-derived macrophages were stimulated with LPS or imiquimod to ligate TLR4 or TLR7, respectively. These stimulants are known to enhance the production of many cytokines other than IFN, including IL-1α, IL-6, IL-12, and TNFα, all of which play critical roles in responses to viruses (46). We observed that downregulation of signaling through the IFNAR during ligation of PRRs resulted in dysregulation of cytokines by macrophages and/or impacted the viability of these cells.
Viability of the macrophages was also assessed since TNFα induced by LPS-mediated stimulation of bone marrow-derived macrophages has been shown to induce apoptosis through an autocrine feedback loop (8). Furthermore, apoptosis of macrophages can cause rapid nuclear localization of cytoplasmic IL-1α, which can suppress inflammation, including reducing production of pro-inflammatory cytokines (7). As such, monitoring survival of macrophages is important when studying their pro-inflammatory responses. This study also provided a cell-based model for the discovery of molecular mechanisms controlling cytokine responses to facilitate the identification of strategies to modulate inflammation and cytokines during viral infections.
Materials and Methods
Mice
The Animal Care Committee at the University of Guelph approved all procedures involving mice (animal utilization protocol #3807), which conformed to the guidelines and policies of the Canadian Council on Animal Care. Wild-type C57BL/6 mice (strain No. 027; Charles River Laboratories) were housed at the Ontario Veterinary College Isolation Unit, University of Guelph, and were donors of bone marrow to facilitate the culturing of macrophages.
Macrophage cultures
Mouse-derived tibias and femurs were the primary sources of bone marrow. Single-cell suspensions were prepared before lysis of red blood cells with ACK lysing buffer (Fisher Scientific, Nepean, Canada). The cells were then differentiated into macrophages in complete RPMI-1640 media (Fisher Scientific) supplemented with 10% heat-inactivated bovine calf serum (Fisher Scientific) penicillin/streptomycin (Fisher Scientific) and
Blocking IFNARs
To block signaling through the IFNAR, cells were treated with anti-mouse IFNα/β receptor (Cat. No. I-401; Leinco Technologies, St. Louis, MO) at a concentration of 10 μg/mL for 1 h before any downstream treatments. This antibody binds and blocks IFNAR without causing downstream signaling. To find the optimal length of antibody pretreatment, a time-course experiment was performed, with lengths, including 1, 12, 24, 36, and 48 h (data not shown). As no significant difference was observed between these time points, a 1-h pretreatment was deemed sufficient for IFNAR blockade. Inhibition of signaling through the IFNAR was verified by western blot analysis.
Western blotting
Initial optimization of antibody-mediated blocking of signaling through the IFNAR was performed with an immortalized antigen-presenting cell line (DC2.4s, kindly provided by Dr. Yonghong Wan, McMaster Immunology Research Centre, Hamilton, Canada). Blockade of signaling was assessed by western blotting. Cells were cultured in complete RPMI-1640 medium as described above.
To analyze inhibition of signaling through the IFNAR, western blot analysis showing a reduction of phosphorylated signal transducer and activator of transcription 1 (STAT1) in these DC2.4s stimulated with recombinant IFNβ (Fisher Scientific) was performed. Proteins were harvested from DC2.4 cells by resuspending the cells on ice in radioimmunoprecipitation assay buffer supplemented with sodium orthovanadate, phenylmethanesulfonyl fluoride, and protease/phosphatase inhibitor (Cat. No. P2714-1BTL; Sigma-Aldrich, Oakville, Canada). The mixture was incubated on ice for 10 min and then centrifuged at 14,000 g for 15 min at 4°C to separate cell debris from proteins. The supernatant was collected and kept at −20°C until quantification with a Bradford assay.
To quantify the proteins in cell lysates for western blotting, a Bradford assay was performed. Seven protein standards were prepared fresh with 1 μg/μL of bovine serum albumin (Cat. No. BP-1600-1; Fisher Scientific), DNAse/RNAse-free water (Cat. No. BP2484-100; Fisher Scientific), and Bradford reagent (Cat. No. 500-0205; Bio-Rad, Mississauga, Canada) to the final concentrations of 0, 1, 5, 10, 15, 25, and 50 μg/mL.
Each standard and sample were plated in duplicate in a 96-well round-bottom plate (Nunc, Cat. No. 12-565-65; Fisher Scientific) (200 μL per well). Absorbance was then measured on a plate reader (GloMax Multi-detection system; Promega, Madison, WI) at a wavelength of 595 nm. The absorbance values of standards were plotted versus their known protein concentrations to obtain a trend line and an associated equation. This equation was then used to determine the protein concentration of each test sample and, thus, the required volume to provide equal amounts of proteins in western blots.
Prepared samples were pulsed with 6 × loading buffer (supplemented with 2-mercaptoethanol). Samples were subsequently placed on a heat block at 90°C for 5 min, then vortexed. Samples were run immediately or stored at −20°C for analysis at a later date. For analysis, prepared samples and a ladder (Cat. No. PM005-0500, PiNK Plus Prestained Protein Ladder; FroggaBio, Toronto, Canada) were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis at 120–130 V for 1–1.5 h.
After electrophoresis, proteins were transferred to polyvinylidene fluoride membranes (Cat. No. CA28148-752; VWR, Mississauga, Canada) at 100 V for 1 h, before being blocked for 1 h in a buffer (5% skim milk in 1 × Tris-buffered saline [Cat. No. BP-2471-1; Fisher Scientific] + 250 μL of Tween20 [Cat. No. 194841; MP Biomedicals, Solon, OH]; TBST). The two primary antibodies used for detection were phospho-STAT-1 rabbit mAb (Y701) (Cat. No. 7649L; Cell Signaling Technology) and native STAT1 rabbit mAb (Cat. No. 9175L; Cell Signaling Technology) to measure levels of phosphorylated STAT1 and native STAT1 in the same samples, respectively. STAT1 is downstream in the signaling pathway following ligation of IFNAR.
Membranes were washed in TBST and rocked for 1 h at room temperature while being stained with a secondary antibody (Cat. No. 621040, horseradish peroxidase-conjugated goat anti-rabbit IgG; Life Technologies, Burlington, Canada), which was washed off with TBST before imaging. To obtain images, membranes were placed on an overhead sheet and incubated with enhanced chemiluminescence solution (Cat. No. WBLUF0100, Luminata Forte Western HRP Substrate; Millipore Corp., Billerica, MA) for 1 min before their image was recorded using a ChemiDoc system (Bio-Rad).
To show that equal quantities of protein were loaded, membranes were reblocked and stained using an actin-specific antibody (Cat. No. sc-1616; Actin I-19, goat polyclonal IgG; Santa Cruz Biotechnology, Dallas, TX) at a dilution of 1:2,000 in blocking buffer overnight at 4°C. The rest of the protocol was carried out as per the western blotting procedure described above. The secondary antibody to detect actin was horseradish peroxidase-conjugated rabbit anti-goat IgG (H+L) (Cat. No. 611620; Invitrogen, Frederick, MO) diluted 1:10,000 in 5% skim milk.
Flow cytometry
Before staining surface markers, macrophages were resuspended in anti-CD16/32 to block Fc receptors (BD Biosciences, San Diego, CA). CD11c-PE-Cy7 (Cat. No. 25-0115-82; eBioscience, San Diego, CA), CD11b-eFluor450 (Cat. No. 48-0112-82; eBioscience), F4/80-FITC (Cat. No. 11-4801-85; eBioscience), CD45RA-biotin (Cat. No. 557460; BD Biosciences, Mississauga, Canada), and Streptavidin-APC-Cy7 (Cat. No. 554063; BD Biosciences) were used to phenotypically characterize macrophages.
After the phenotypic staining described above, cells were washed in phosphate-buffered saline and incubated with a fixable viability dye (Cat. No. 65-0865-18, Fixable Viability eFluor-780; eBioscience). Cells were then fixed and permeabilized with Cytofix/Cytoperm (Cat. No. 555028; BD Biosciences). Fixed and permeabilized cells were stained with cytokine-specific antibodies; TNFα-PE (Cat. No. 12-7321-82; eBioscience), IL-12-eFluor660 (Cat. No. 50-7123-82; eBioscience), IL-6-APC (Cat. No. 561367; BD Biosciences), and IL-1α-PE (Cat. No. 12-7001-82; eBioscience). Samples were filtered with nylon mesh (Cat. No. 03-48/31; Elko Filtering Co., Switzerland) and then analyzed using a three-laser FACSCanto II flow cytometer (BD Biosciences, Mississauga, Canada).
Kinetic assessment of cytokine production by cells
To evaluate the changes of cytokine production in macrophages over time, 60-h time course experiments were conducted. Macrophages were differentiated from bone marrow and plated. On day 6 of differentiation, half of the samples were given a 1-h pretreatment with IFNAR-blocking antibody, then either left unstimulated or stimulated with LPS or imiquimod (2 and 1 μg/mL, respectively) (Sigma-Aldrich) for 5 h; this represented “time zero” for the time-course experiment. Brefeldin-A was added 12 h before harvesting macrophages through cell scraping for phenotypic and intracellular cytokine staining. Both the percentage of cells producing a cytokine and the amount of cytokine produced per cell (mean fluorescence intensity) were determined using FlowJo version10 (BD Biosciences) and graphed using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA).
Trypan blue dye exclusion test
To quantify live versus dead cells, 10 μL of cell suspension was mixed with 10 μL of trypan blue dye (Life Technologies) then counted using an automated TC-20 cell counter (Bio-Rad).
Data analysis and statistics
Analysis of flow cytometric data, including determination of areas under the curves, was done using FlowJo version 10 Single Cell Analysis Software (Tree Star, Inc., San Carlos, CA). Untreated background levels for the proportion of cells producing cytokines were subtracted from all samples for each replicate. Means and standard errors were graphed using GraphPad Prism version 8 and analyzed by one-way (single time point experiments) or two-way (time-course experiments) analysis of variance, with statistical significance being defined as a p-value <0.05.
Results
Anti-mouse IFNAR blocked IFNβ-induced Stat1 phosphorylation in myeloid cells
The DC2.4 cell line was initially used to confirm the effectiveness of the IFNAR-blocking antibody. The cells were stimulated with IFNβ following incubation with the IFNAR-blocking antibody, and then phospho-STAT1 levels were assessed by western blotting. Compared to IFNβ-stimulated cells with functional IFNAR, less phospho-STAT1 was detected in the IFNAR-blocked sample, indicating successful blockage of downstream pathways (Fig. 1A). In contrast the amount of native STAT1 was unchanged (Fig. 1B). Furthermore, the protein loading control suggested that comparable levels of proteins were loaded into the gel (Fig. 1C). This confirmed the ability of anti-IFNAR to block type I IFN-mediated signaling, thereby providing a model to allow in vitro manipulation of this signaling pathway in myeloid cells.

Anti-mouse IFNAR blocked IFNβ-induced STAT1 phosphorylation in the DC2.4 myeloid cell line. Murine DC2.4 cells were pretreated with an IFNAR-blocking antibody (Ab) or isotype control before being stimulated with recombinant IFNβ. A western blot was performed to compare levels of
Differential cytokine generation was demonstrated in the absence of IFNAR signaling upon ligation of TLR4 or TLR7
The ability to block the IFNAR allowed analysis of the role of this signaling pathway in cytokine production during the stimulation of either TLR4 or TLR7 on/in macrophages. The proportion of macrophages (as defined in Supplementary Fig. S1) producing TNFα, IL-12, IL-6, and IL-1α within the first 12 h after stimulation was measured and plotted after removing the mean background staining of untreated control cells. IFNAR blockade impacted cytokine production by macrophages. The proportion of macrophages producing TNFα, IL-12, and IL-6 was significantly increased in the absence of IFNAR signaling during TLR4 ligation (Fig. 2). In contrast, in the macrophages lacking functional IFNAR signaling, TLR7 stimulation had no impact on the proportion producing TNFα and IL-12 and led to a small but significant decrease in the production of IL-6 and a larger decrease for IL-1α (Fig. 3).
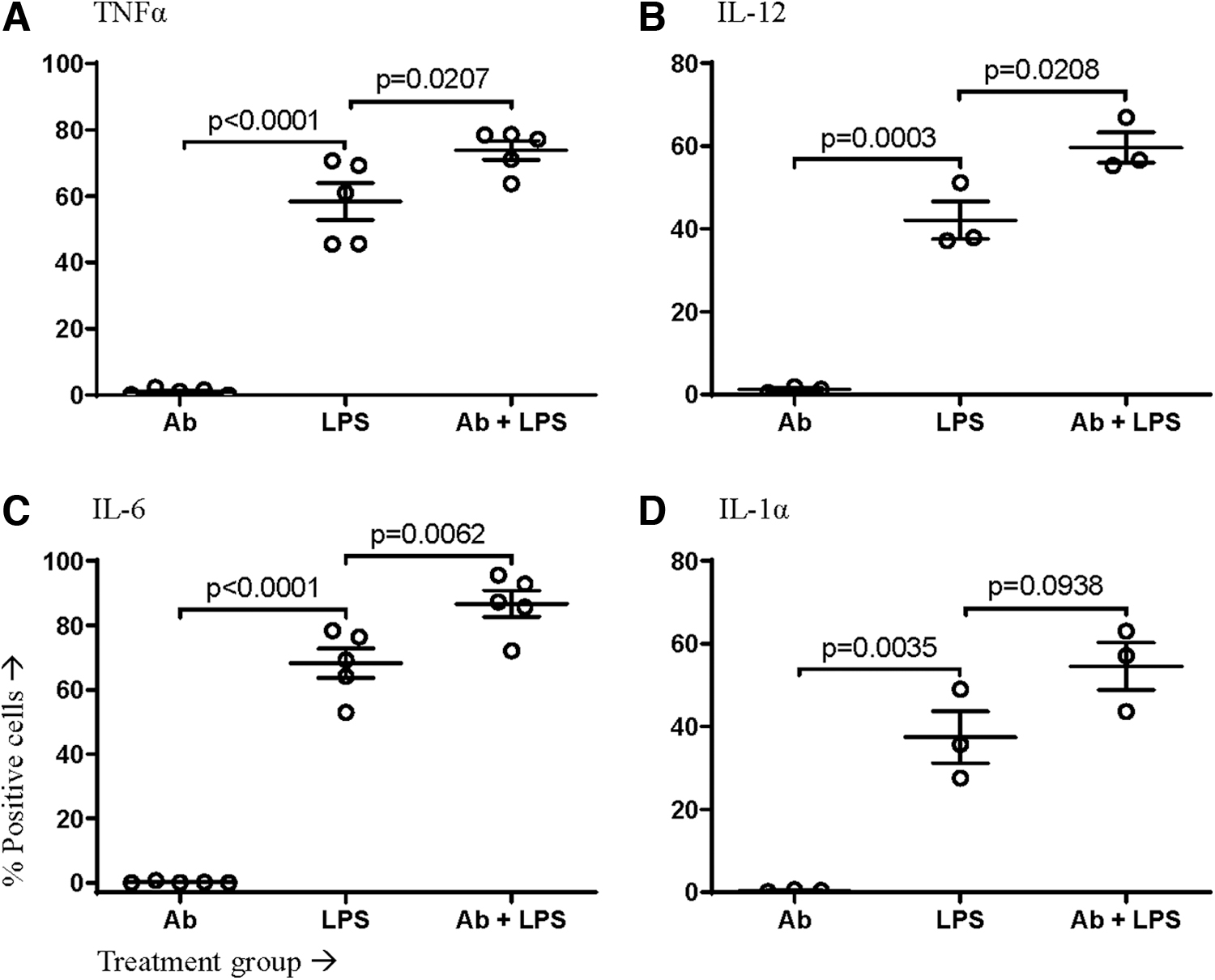
IFNAR blockade caused macrophages to produce more inflammatory cytokines in response to LPS. Murine macrophages differentiated from bone marrow-derived progenitor cells were pretreated with an IFNAR-blocking antibody (Ab) or an isotype control. Cells were then left unstimulated or stimulated with LPS. The proportion of viable macrophages producing the cytokines
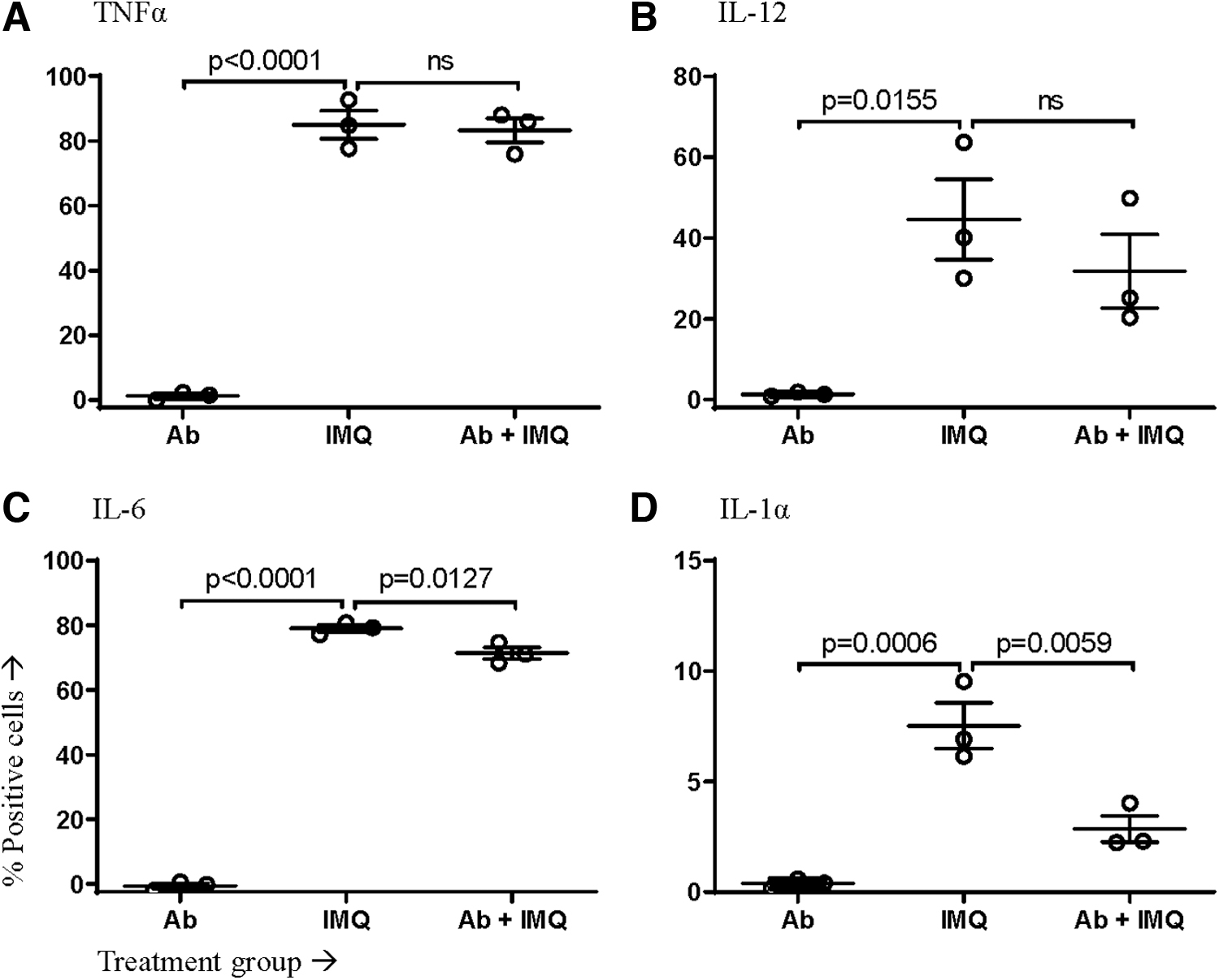
IMQ-stimulated macrophages produced less cytokines during IFNAR blockade. Murine macrophages differentiated from bone marrow-derived progenitor cells were pretreated with IFNAR-blocking antibody (Ab) or an isotype control. Cells were then left unstimulated or stimulated with IMQ. The proportion of viable macrophages producing the cytokines
Kinetics of cytokine production differed in macrophages with and without IFNAR signaling
After observing the production of cytokines by macrophages during the first 12-h window after administration of brefeldin-A, we sought to see if these changes were consistent up to 60 h poststimulation. To observe these changes over time, macrophages were stimulated, with half of the cells being pretreated with the IFNAR-blocking antibody. Samples were serially collected every 12 h until all five windows of time up to 60-h poststimulation were collected. Brefeldin-A was added to each sample 12 h before collection.
In addition to looking at the proportion of cells producing cytokines, we also analyzed the mean fluorescence intensity for each cytokine as a surrogate measure of the relative amount of the cytokine being produced on a per-cell basis. The proportion of LPS-stimulated macrophages producing cytokines was elevated during the first 12 h when IFNAR signaling was blocked; this held true for all four cytokines (Fig. 4). This increase extended to 24 h for IL-6 (Fig. 4C). TNFα and IL-6 returned to baseline levels by 36 h poststimulation (Fig. 4A, C).
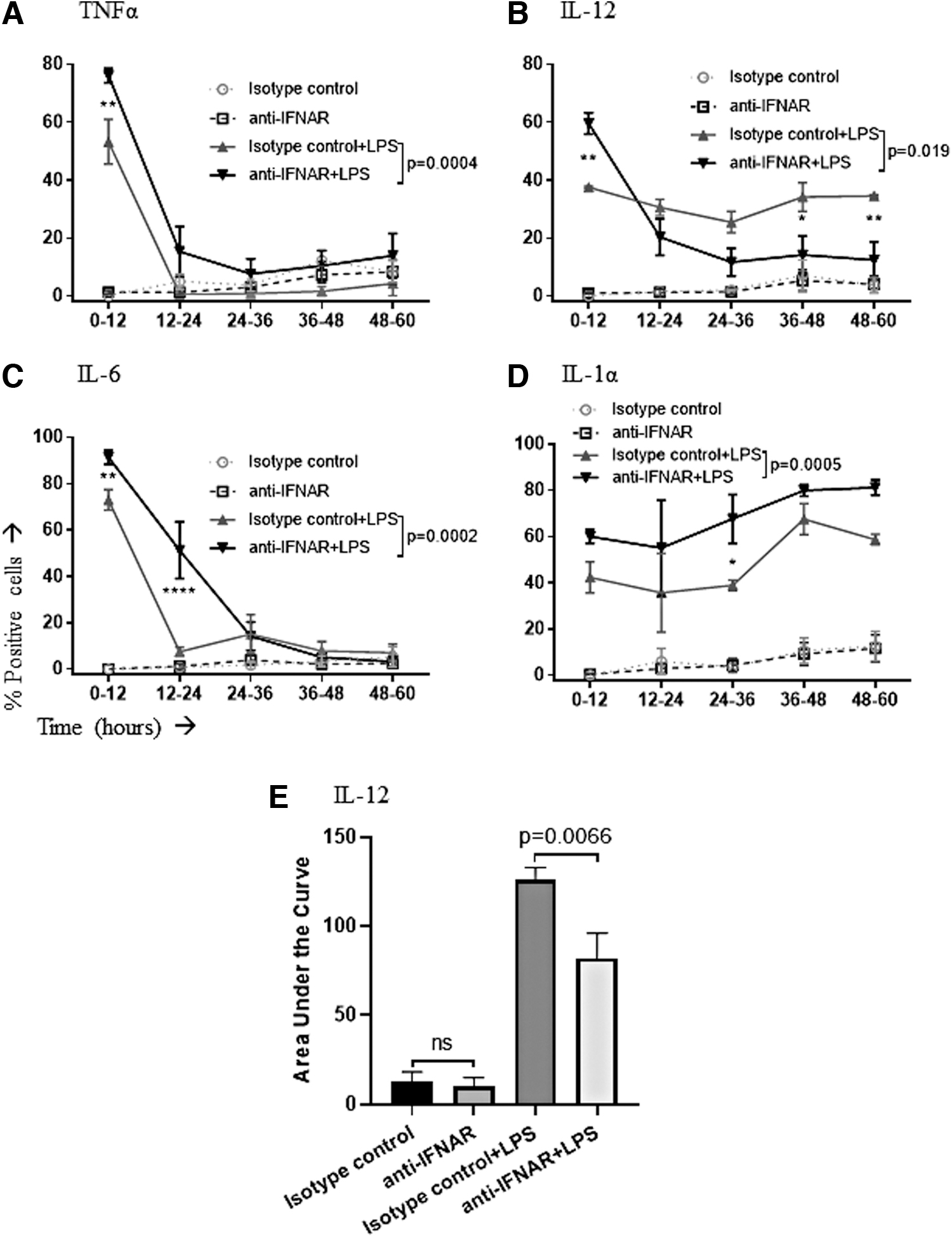
Time course of cytokine production by LPS-stimulated macrophages when IFNAR-mediated signaling was disrupted. Macrophages were pretreated with an IFNAR-blocking antibody (anti-IFNAR) or an isotype control. Cells were then left unstimulated or stimulated with LPS. The proportion of viable macrophages producing the cytokines
IFNAR-blocked cells showed an initial upregulation in IL-12 production before dropping below the cells with functional IFNAR after the first 12 h (Fig. 4B). Analysis of areas under these curves revealed that the net effect was an overall reduction in the total proportion of IL-12-producing macrophages over a period of 60 h (Fig. 4E). A greater proportion of cells produced IL-1α at all time points with IFNAR blockade (Fig. 4D). However, the amounts of cytokines being produced per cell were unchanged for all four cytokines (Fig. 5).
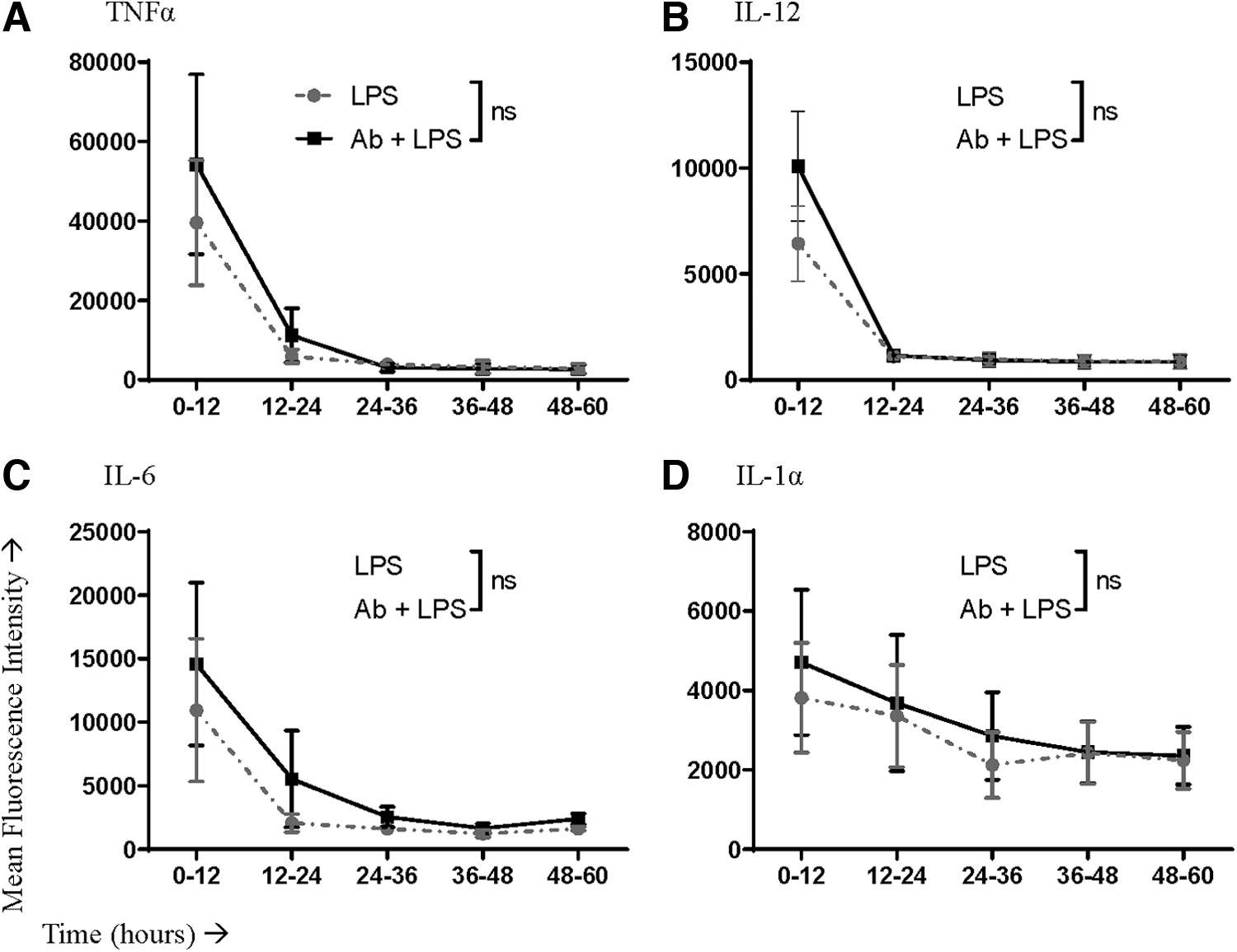
Mean fluorescence intensity values of antibody staining for cytokine detection in LPS-stimulated macrophages with or without blocking the IFNAR. Macrophages were pretreated with an IFNAR-blocking antibody or an isotype control. Cells were then left unstimulated or stimulated with LPS. Macrophages producing cytokines within consecutive 12-h windows poststimulation were assessed by flow cytometry to determine the relative amount of the cytokine being produced per cell based on the mean fluorescence intensity. Cytokines that were evaluated included
Unlike LPS stimulation, macrophages stimulated with imiquimod had no change in the proportion producing TNFα or IL-12 at any time point (Fig. 6A, B). Furthermore, a lack of IFNAR signaling reduced the proportion of macrophages producing IL-6 and IL-1α within the first 12 h of responding to imiquimod (Fig. 6C, D). These reductions were relatively small, and IFNAR-mediated differences were no longer apparent beyond 12 h poststimulation. Similar to LPS stimulation, IFNAR blockade seemed to have no significant impact on the amounts of cytokines produced per macrophage in response to imiquimod-mediated stimulation (Fig. 7).

Time course of cytokine production by IMQ-stimulated macrophages with or without IFNAR blockade. Macrophages were pretreated with an IFNAR-blocking antibody (anti-IFNAR) or an isotype control. Cells were then left unstimulated or stimulated with IMQ. The proportion of viable macrophages producing the cytokines
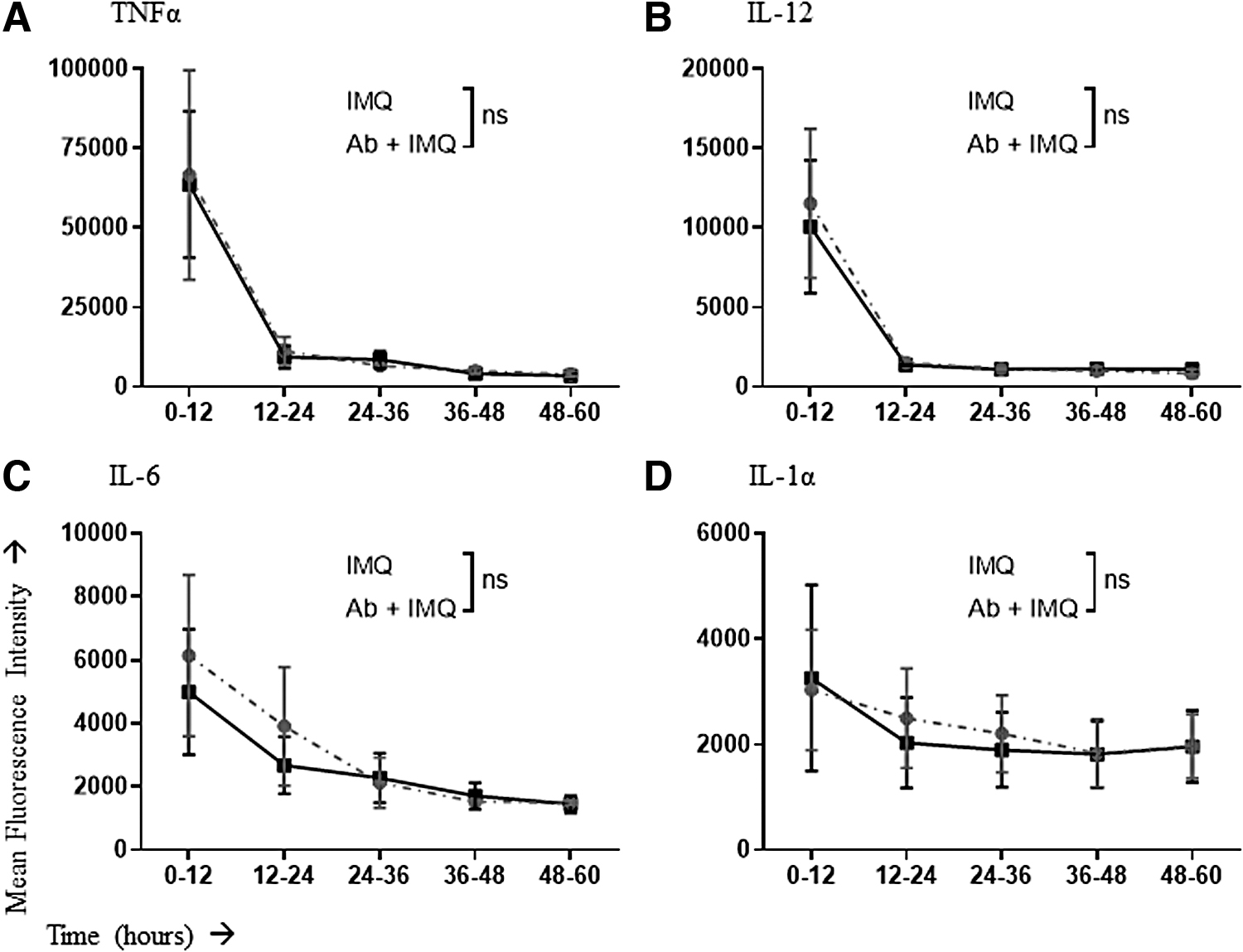
Mean fluorescence intensity values of antibody staining for cytokine detection in IMQ-stimulated macrophages with or without IFNAR blockade. Macrophages were pretreated with an IFNAR-blocking antibody or isotype control. Cells were then left unstimulated or stimulated with IMQ. Macrophages producing cytokines within consecutive 12-h windows poststimulation were assessed by flow cytometry to determine the relative amount of the cytokine being produced per cell based on the mean fluorescence intensity. Cytokines that were evaluated included
IFNAR blockade appeared to have a protective effect on the survival of macrophages stimulated with LPS but promoted death if stimulated with imiquimod
Having shown that disruption of IFNAR signaling led to an increase in the proportion of macrophages producing cytokines in response to stimulation with LPS, we further asked whether the presence or absence of IFNAR signaling was affecting the absolute number of cells producing cytokines. To do this, equal numbers of macrophages (i.e., 6 million) were plated. Some were pretreated with anti-IFNAR, others were not. Then cells were either left unstimulated or stimulated with LPS or imiquimod. Twenty-four-, 48-, and 72-h later, live cells (as determined by trypan blue dye exclusion) were enumerated. Interestingly, IFNAR blockade resulted in there being almost threefold more macrophages 72-h poststimulation with LPS compared to cells with intact IFNAR signaling (Fig. 8A).
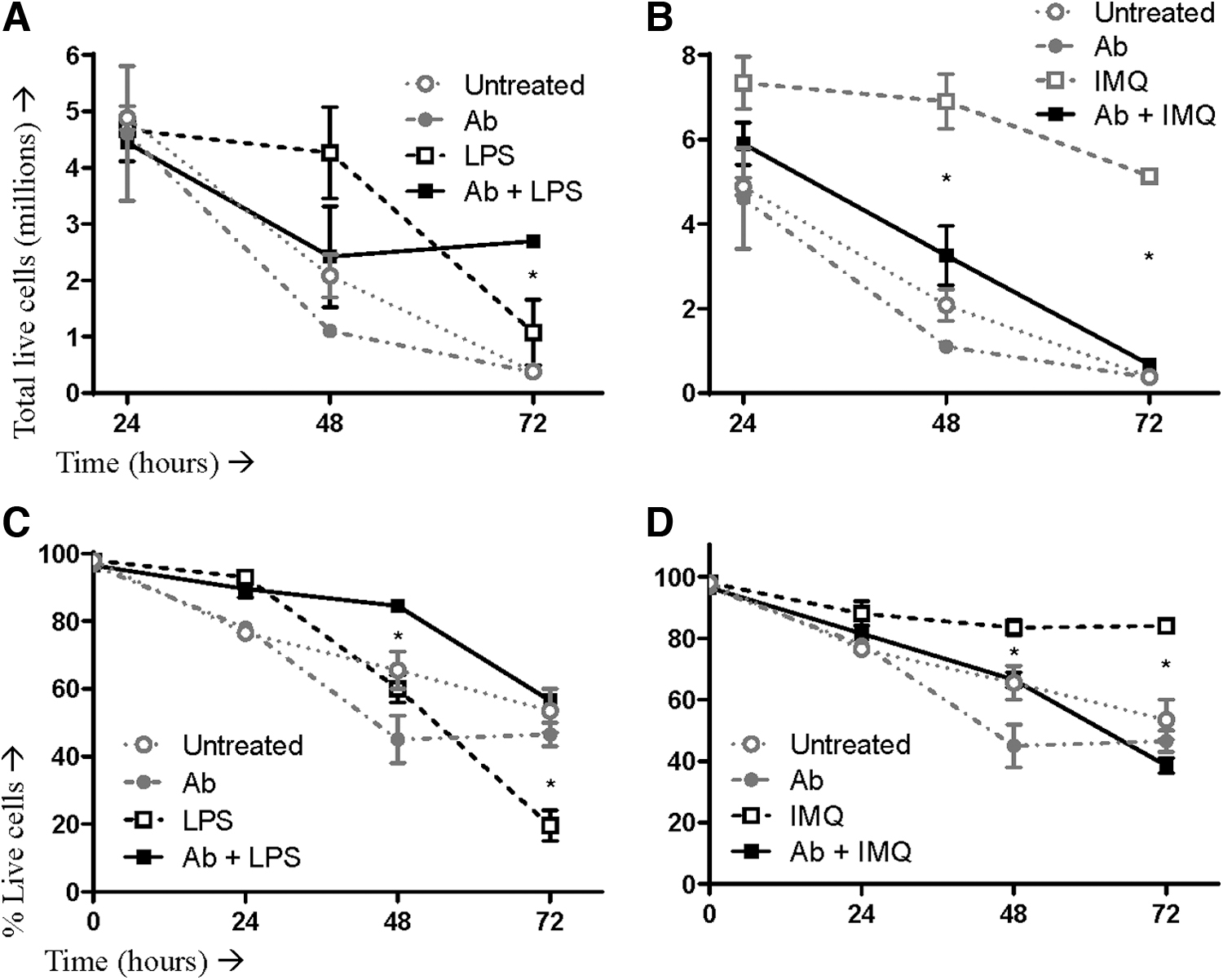
Survival of LPS- or IMQ-stimulated macrophages when type I IFN signaling was left intact or disrupted. Murine bone marrow-derived macrophage cultures were split in two, with half treated with an IFNα/β receptor-blocking antibody (Ab) and half left untreated. Cells were then left unstimulated or stimulated with LPS or IMQ. Cells were initially seeded at 6 million per well and then harvested at 24, 48, and 72-h poststimulation. At each time point, 10 μL of cells were mixed with 10 μL of trypan blue dye and counted using an automated cell counter. Shown are absolute counts of live macrophages after stimulation with
This effect was reversed for imiquimod, with IFNAR blockade potentiating more than a 10-fold reduction in the number of macrophages 72 h poststimulation (Fig. 8B). These results correlated with an enhanced viability of macrophages when they were stimulated with LPS during concomitant IFNAR blockade but a reduced viability when IFNARs were blocked during imiquimod-mediated stimulation (Fig. 8C, D).
Discussion
In this study, the production of inflammatory cytokines and whether these could be manipulated by way of IFNAR signaling during either TLR4 or TLR7 stimulation in macrophages was evaluated. Bone marrow-derived macrophages were studied since evidence suggested that while all nucleated cells have IFNAR signaling capabilities, it is cells of the myeloid lineage that are primarily responsible for the antiviral effects of type I IFN signaling in vivo (38).
Due to the potent antiviral properties these IFN signaling pathways promote, it is wholly unsurprising that mice lacking IFNAR signaling succumb to viral infections more rapidly than wild-type mice (e.g., Yellow fever virus and Ebola virus) (47). Pinto et al. demonstrated increased fatality in IFNAR−/− mice after infection with West Nile Virus, not due to viral infection per se, but due to a cytokine storm brought on by the dysregulation of the pro-inflammatory cytokine TNFα.
Furthermore, they also designated the mitochondrial antiviral signaling protein (MAVS), in the retinoic acid-inducible gene (RIG)-I pathway, as the key molecule involved in cytokine production and eventual sepsis during West Nile virus (WNV) infection. They observed that in mice lacking both IFNAR and MAVS (double knockout), cytokine production was completely dampened even with robust WNV infection, compared to both wild type and IFNAR−/− (single knockout) (38).
This role of IFNAR and downstream components in the contribution of cytokine production is likely not unique to WNV and RIG-I activation. For example, STAT1 deficiency led to similar results during infection with Listeria monocytogenes (Gram-positive bacteria, sensed by TLR2) (22). Thus, the study presented here focused on the effects resulting from ligation of two different PRRs: TLR4 and TLR7. These were chosen, in part, due to their involvement in instigating immune responses to a variety of stimuli, including both viral and bacterial pathogens and mimics.
In the present study, following blockage of the IFNAR pathway, TLR4 stimulation led to a significant acute increase in the proportion of macrophages producing TNFα, IL-12, IL-6, and IL-1α within the first 12 h after PRR ligation. In contrast, ligation of TLR7 resulted in a decrease in the proportion of cytokine-producing cells when functional IFNAR signaling was impaired.
Specifically, the percentage of macrophages producing IL-6 and IL-1α was reduced. With that said the relative change in these percentages was small, bringing into question whether it would be biologically significant. It is notable that IFNAR blockade during stimulation with PRR agonists modulated the frequency of cells producing cytokines but not the relative amounts of cytokines being produced per cell. This suggests that interference with IFNAR signaling sensitized otherwise nonresponsive macrophages to begin producing pro-inflammatory cytokines when stimulated with LPS, but the opposite effect occurred with imiquimod.
TNFα is thought to be the biggest contributor in the induction of apoptosis (4) and uses this mechanism as a method to help protect the host from viral infection (50). However, increased concentrations of TNFα have been associated with a wide variety of diseases such as arthritis, inflammatory bowel disease (4), chronic heart failure (27), and Alzheimer's disease (10). Thus, anti-TNF therapy has been studied extensively and been implemented into many treatment regimens (32).
In contrast, while there may not be many diseases associated with low levels of TNFα, the ability to downregulate TNFα levels could be beneficial where traditional anti-TNFα therapies fall short. For example, patients with rheumatoid arthritis have been shown to be at an increased risk for serious infections (48). Furthermore, in the context of some diseases such as Sjogren's Syndrome, anti-TNFα had absolutely no benefit above the placebo, as shown in a randomized, double-blind study carried out by Mariette et al. (29).
Even worse, due to the induction of antinuclear antibodies, some TNFα-inhibitor treated patients actually develop new autoimmune pathologies, including drug-induced lupus (41). Quite simply, blocking the production of specific cytokines pharmacologically can have profound effects in the reduction of inflammation mediated by the immune system (26). However, there are also many associated risks that warrant the further investigation into ways to improve these therapies.
In the context of TLR7 stimulation, our results showed that the proportion of macrophages producing TNFα was unaffected within the first 12 h when signaling through the IFNAR was blocked. However, the number of IFNAR-blocked macrophages producing TNFα decreased disproportionately relative to those with intact IFNAR signaling at 48- and 72-h after stimulation with imiquimod (Fig. 8B). This would imply that concomitant treatment with anti-IFNAR and imiquimod could be a strategy to rapidly deplete TNFα-producing macrophages. It would be interesting to investigate the potential for additive or synergistic effects of combining this approach with anti-TNFα therapy (13).
Upregulation of IL-12 may not carry the same negative connotations as increased levels of TNFα. IL-12 plays a strong role in directing naive T cells to T helper-1 (Th1) CD4+ T cells, which is important for resolution of many diseases (45). The Th1 response facilitates cell-mediated immunity, which is crucial for adequate defense against viral and other intracellular pathogens270, as opposed to the antibody-mediated Th2 response (42).
It is well documented that the development of a given T helper response is important as some pathogens are more effectively cleared by one or the other, so the ability of IL-12 to direct this immune response is important, especially in viral infections (45). In our study, when macrophages were undergoing stimulation with LPS, the proportion producing IL-12 was initially increased with IFNAR blockade. This then decreased relative to cells with intact IFNAR signaling at later time points, resulting in an overall decrease in the average proportion of macrophages producing IL-12 for 60-h post-stimulation, as measured by areas under the curves (p = 0.066, Fig. 4B, E).
However, this appeared to be offset by enhanced survival of LPS-stimulated macrophages with IFNAR blockade (Fig. 8A). We speculate that this may have been due to reduced sensitization of the macrophages to type I IFN-induced apoptosis (44). Although the proportion of IL-12-producing macrophages was unchanged during TLR7 stimulation with IFNAR blockade, the numbers of these cells dropped precipitously over 72 h. Due to the strong antiviral properties of IFNs, it is counterintuitive to consider blocking IFNAR as an antiviral measure. In fact, groups have shown viral infections to be more lethal in IFNAR−/− mice [Karimi K and Bridle B, personal observations and (3)]. However, if IFNAR blockade could be combined with TLR4 ligation it might lead to an upregulation of IL-12, which could potentially help steer the immune system toward a Th1 response, which is the desired type for viral pathogens.
Increased concentrations of IL-6 have been implicated in many issues faced by sufferers of chronic inflammation (e.g., arthritis). It plays a fundamental role in directing acutely activated inflammatory mechanisms to a chronic state, including the accumulation of mononuclear cells at sites of injury and antiapoptotic effects on T cells, among others (18). Specifically in rheumatoid arthritis, IL-6 has a pivotal position in joint inflammation due to the high bioactivity of IL-6 in synovial fluid and synoviocytes (19).
Our study showed an increased proportion of IL-6-producing macrophages during simultaneous IFNAR blockage and TLR4 stimulation. This could provide novel insight into the underlying mechanisms of the poorly understood onset of diseases such as rheumatoid arthritis. The development of autoimmune diseases and chronic inflammation can take on a wide range of devastating and debilitating clinical presentations, so the control of IL-6 is an attractive target when considering the control of cytokine production.
Our demonstrated ability to dampen the production of this cytokine during TLR7 stimulation in the absence of IFNAR signaling could be explored as a therapeutic mechanism in the future. However, IL-6 is complex. Not only is it regarded for its pro-inflammatory properties, but it can also play a role in anti-inflammatory processes and regeneration (19). The regulation of IL-6 is often therapeutically addressed by targeting the receptors (18), but the differences observed during different TLR stimulations could have an impact on the outcomes of these therapies and, as such, should be investigated further.
IL-1α is a pro-inflammatory cytokine and as such plays a role in the induction of inflammation, fever, and sepsis. Interestingly, IL-1α can be both secreted and surface expressed, and both versions are bioactive (11). The flow cytometry-based assay used in this study could detect both forms of IL-1α. Its role of IL-1α in inflammation has been demonstrated as being crucial in the progression and development of arthritic diseases such as rheumatoid arthritis. It has been demonstrated that collagen-induced arthritis in mice could develop following systemic administration of IL-1 (35) and the development of collagen-induced arthritis can also be seen following systemic administration of LPS, which stimulates TLR4 (16). This is not altogether surprising, given that LPS activates macrophages, and IL-1α is secreted by activated macrophages. However, these processes are yet to be examined in the context of signaling through the IFNAR.
In this study, we saw a significant increase in the proportion of LPS-stimulated macrophages producing IL-1α across the entire 60-h duration of the experiment (p = 0.0005, Fig. 4D), as well as promotion of their survival (p < 0.05, Fig. 8A), in the absence of IFNAR signaling. This association poses the question, then, if an increase of IFNAR signaling could drive the production of IL-1α back down and thus improve the outcome of anti-IL-1 treatment in the context of inflammatory diseases like rheumatoid arthritis. Similar to anti-TNF treatment, anti-IL-1 treatment has been used to counteract inflammatory joint diseases (31).
Anti-IL-1 treatment was shown by Joosten et al. to prevent rheumatoid arthritis prophylactically and substantially ameliorate disease progression in patients with established rheumatoid arthritis. However, novel methods for the improvement of treatments are sought after (20). Due to our observed significant decrease in IL-1α during TLR7 stimulation in the absence of IFNAR signaling, the possibility of combining this with anti-IL-1 should be investigated, as with the anti-TNFα discussed above.
The assay for enumerating cell numbers and viability yielded some notable results (Fig. 8). Specifically, IFNAR signaling appeared to accelerate the death of macrophages activated by LPS. In stark contrast, intact IFNAR signaling during treatment with imiquimod dramatically enhanced the survival of macrophages. This could have important implications in the context of a variety of diseases, including those caused by infectious agents.
Notably, microbes that modulate IFNAR signaling pathways could potentially control the fate of macrophages. If this were done in the context of dominant signaling through PRRs such as TLR7, it could promote survival of macrophages and possibly be an important mechanism in the life cycle of macrophage-tropic microorganisms. Alternatively, impairing IFNAR signaling while ligating TLR4 could be a potential escape strategy for pathogens by promoting the depletion of macrophages.
In this study, we found that IFNAR-mediated signaling could act as either a negative or a positive regulator of a network of inflammatory cytokines when accompanied by simultaneous TLR stimulation. As such, this pathway can be manipulated to restore homeostasis during excessive inflammation and cytokine storms. This ability to further study and characterize the IFNAR pathway and its role in cytokine regulation could allow for the determination of novel targets for the suppression of inflammation, which could be important for moderating the bystander tissue damage often observed in many nonlife threatening diseases.
In addition, the ability to manipulate cytokine levels could prove paramount during cytokine storms, such as those associated with septic shock or viremia. Furthermore, both inflammation and cytokine dysregulation are associated with a huge array of clinical situations, so this added method of control could prove impactful in a wide variety of pathologies.
In summary, signaling through the IFNAR on macrophages appears to regulate a network of cytokines and could potentially be manipulated to restore homeostasis during excessive inflammation and/or cytokine storms. As cytokine storms have the potential for toxicity and have been associated with many different pathologies (e.g., acute lung damage associated with avian H5N1 influenza virus, severe acute respiratory syndrome from coronaviruses, bacteria like Staphylococcus aureus, multiple sclerosis, graft-versus-host disease, and even consequences from various therapies), the need for a greater understanding of the underlying mechanisms is needed. Furthermore, manipulation of IFNAR signaling during either TLR4 or TLR7 stimulation of macrophages could be a strategy to optimize cytokine profiles to support the elimination of viral and bacterial pathogens.
Footnotes
Authors' Contributions
Conceptualization, A.S.R., S.R.W., A.M.V.-P., S.K.W., B.W.B., and K.K.; Data curation, A.S.R., S.R.W., L.C., and B.W.B.; Formal analysis, A.S.R., S.R.W., L.C., A.M.V.-P., S.K.W., B.W.B., and K.K.; Funding acquisition, B.W.B.; Investigation, A.S.R., S.R.W., L.C., and B.W.B.; Methodology, A.S.R., S.R.W., and B.W.B.; Project administration, B.W.B.; Resources, B.W.B.; Supervision, A.M.V.-P., S.K.W., B.W.B., and K.K.
Acknowledgments
The authors thank Campus Animal Facilities, University of Guelph, for animal care services. This work was based, in part, on research conducted for a thesis written by A.S.R. This thesis can be found in the following online repository:
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Operating funds were from a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC; awarded to BWB; Grant No. 436264-2013). Stipend funding for highly qualified personnel was provided by an Ontario Veterinary College Graduate Scholarship (for ASR) and a NSERC Undergraduate Student Research Award (for L.C.).
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.