
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) is the pathogen of the porcine reproductive and respiratory syndrome, which is one of the most economically devastating diseases of the swine industry. However, whether the inactivated vaccine and modified live attenuated vaccines are effective in disease control is still controversial. Although several groups developed PRRSV virus-like particles (VLPs) as a vaccine against PRRSV, all these VLP-based vaccines targeted PRRSV-2, but not PRRSV-1 or both. Therefore, it is urgent to produce VLPs against PRRSV-1. In this study, we rescued recombinant baculovirus expressing GP5 and M proteins of PRRSV-1 through the Bac-to-Bac® baculovirus expression system. Thereafter, PRRSV VLP was obtained efficiently in the recombinant baculovirus-infected High Five insect cells. Moreover, the PRRSV VLP and PRRSV VLP+A5 could efficiently trigger specific humoral immune responses and B cellular immune responses through intranasal immunization. The combination of PRRSV VLP and A5 adjuvant could improve the level of the immune response. The PRRSV-1 VLPs generated in this study have greater potential for vaccine development to control PRRSV-1 infection.
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) is the etiologic agent of the porcine reproductive and respiratory syndrome (PRRS), which has been considered as one of the most economically devastating diseases of the swine industry (3,26). Swine is the only host of PRRSV, which can be infected by a variety of exposure routes, including parenteral, intranasal, intramuscular, oral, intrauterine, and vaginal, resulting in severe respiratory and reproductive disease in pigs with poor performance and high mortality (1,19). Once infected, the virus replicates in monocytes, including porcine alveolar macrophage and dendritic cells, and the infected animal sheds the virus through saliva, nasal secretions, urine, semen, mammary secretions, and feces (1,19).
PRRSV belongs to the Arteriviridae family, appearing as a 50–60 nm spherical or oval-shaped virion with a relatively smooth surface (19). The viral genome is a single-stranded positive-sense RNA ∼15 kb in length, containing 11 known open reading frames (ORFs) (3,12,19). ORFs 2–7 encode eight structural proteins, including envelope proteins (GP2, E, GP3, GP4, ORF5a, GP5), membrane protein (M), and nucleocapsid protein (N), among which GP5 and M form a disulfide-linked heterodimer and together constitute the major glycoprotein complex on the surface of the virion (3,12,19). Based on the genome sequences, PRRSV was divided into two genotypes: type 1 (European-like or PRRSV-1) and type 2 (North American-like or PRRSV-2) (8,12,24), which are found worldwide. Unexpectedly, although clinical symptoms and genomic organization of two genotypes are similar, the nucleotide acids share only 40–60% sequence identity between different genotypes (8,12,24).
Furthermore, PRRSV-1 was subdivided into four subtypes, including I, II, III, and IV, which mainly circulate in European countries, the United States, Russia, Thailand, and so on (18,20,29,30). In China, PRRSV-2 was the predominant genotype in past decades; however, PRRSV-1 is gradually detected and isolated in recent years (5,13,39).
To control the disease, several kinds of vaccines, including inactivated vaccines and modified live attenuated vaccine (MLV), have been developed. However, whether these vaccines are effective in disease control is still controversial. One reason is that inactivated PRRSV cannot prevent clinical signs and viremia during the virus infection; the other reason is that MLVs only induce partial protection or no protection against heterologous isolates (4,6,23,24,41). Notably, PRRSV displays a high mutation rate, especially due to recombination between MLV strain and wild-type strain reported in recent years, making the development of safe and efficacious PRRSV vaccines a top challenge (3,4,22,24,34).
Virus-like particles (VLPs) are biological nanostructures consisting of viral surface structural proteins assembled in a morphology that mimic the native virion but do not contain viral genetic materials (11,25). Because VLP is replication deficient, it is expected to be a candidate for vaccine and drug delivery (10,11,21,25). Previously, several groups described the generation and evaluation of the PRRSV VLPs containing the viral surface proteins GP5, GP5 and M, or GP5-GP4-GP3-GP2a-M as a vaccine against PRRSV (2,10,23,35,36,38). However, most of these VLP-based vaccines targeted PRRSV-2, but not PRRSV-1 or both. Therefore, considering the extensive use of vaccines based on PRRSV-2 for preventing and controlling PRRS in mainland China, it is urgent to develop safe and efficacious PRRSV vaccines for the PRRSV-1 infection in China.
In this study, we established a reliable convenient method for dual expression of PRRSV-1 proteins (GP5 and M) in the Bac-to-Bac® baculovirus expression system and producing PRRSV-1 VLPs. The immunogenicity of VLP was also evaluated in vitro and in vivo, which suggests that the VLPs obtained in this study would be used for vaccine development against the PRRSV-1 infection.
Materials and Methods
Plasmid construction
GP5 and M genes of PRRSV-1 were amplified from PRRSV LV strain (GenBank ID: M96262.2. kindly provided by Dr. Wenchao Sun) using primer pairs GP5X-F/GP5K-R and MB-F/MH-R (Table 1) and cloned into pMD™-19T simple vector (Takara, Dalian, China) to generate plasmids 19T-GP5 and 19T-M.
Primers Used in the Present Study
Capital letters indicate restriction endonuclease enzyme sites. The Flag sequence was bold and underlined.
To construct the donor plasmid, the GP5 and M genes were digested from plasmids 19T-GP5 and 19T-M by restriction endonuclease enzymes (NEB, Ipswich, MA) and subcloned into pFastBac™ Dual donor plasmid (Invitrogen, Waltham, MA); successively, the resulting plasmid was designated as pFBD-GP5-M (Fig. 1a). The donor plasmid (shuttle plasmid) pFBD-GP5-M was identified by restriction endonuclease enzymes (Kpn I+Xho I and BamH I+Hind III) and sequencing.
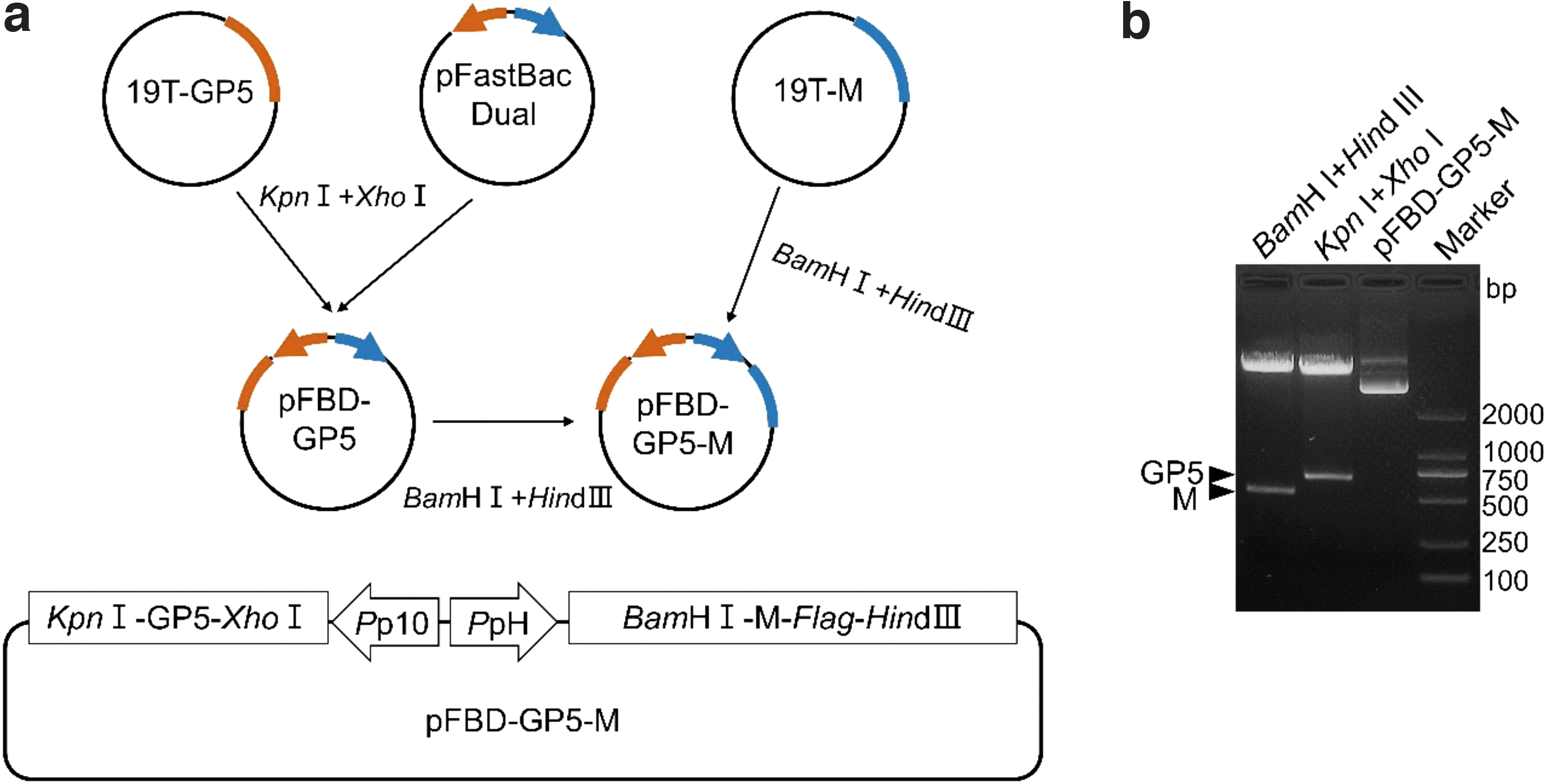
Construction of donor plasmid pFBD-GP5-M.
Preparation of recombinant bacmid
Recombinant bacmid was obtained using the Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer's instruction. Briefly, the donor plasmid pFBD-GP5-M was transformed into Competent DH10Bac™ Escherichia coli cells (Biomed, China) for transposition into the bacmid, followed by the antibiotic selection and blue/white selection. The positive E. coli colonies containing the recombinant bacmid were identified through Polymerase Chain Reaction (PCR) using primer pairs pHF and pUC/M13R, pUC/M13F, and p10R (Table 1). The resulting recombinant bacmid was designated as rBD-GP5-M. The recombinant bacmid DNA was extracted from the E. coli cells and stored at −80°C for further research.
Preparation of recombinant baculovirus
Spodoptera frugiperda (Sf9) cell and High Five insect cells were kindly provided by Prof. Xianzhu Xia. The cells were cultured in SF-900™ II SFM medium (Gibco) supplemented with 10% fetal bovine serum (FBS) and incubated at 27°C.
To rescue the recombinant baculovirus, Sf9 cells were seeded on a six-well plate at a density of 106 cells per well for 30 min. The cells were transfected with 3–4 μg of the bacmid DNA using Cellfectin® II Reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. The supernatants of cells transfected with the recombinant bacmid were collected after culturing for 5–7 days at 27°C and centrifuged at 3,500 rpm, 4°C for 5 min, followed by filtrating with 0.22 μm filter; the resulting recombinant baculovirus was designated as rBDV-GP5-M. Thereafter, the baculoviruses were passaged thrice in Sf9 cells and stored at −80°C. In addition, the viral titer was determined according to the protocol described by BacPAK Baculovirus Rapid Titer Kit (Clontech).
Preparation and purification of PRRSV VLPs
To prepare PRRSV VLPs, High Five cells were infected with recombinant baculovirus rBDV-GP5-M at a multiplicity of infection (MOI) of 4 for 48 h. Cells were collected by low-speed centrifugation (5,000 rpm, 4°C, 5 min) and filtration through a 0.45 μm filter. Then, filtered samples were ultracentrifuged through a 20% sucrose cushion at 35,000 rpm and at 4°C for 2 h. The pellets were resuspended in 500 μL of phosphate buffered saline (PBS). Thereafter, the second ultracentrifugation was performed through a stepwise sucrose density gradient consisting of 1 mL of 20%, 35%, 45%, and 60% sucrose in PBS and centrifuged at 30,000 rpm and 4°C for 2 h. Finally, the purified VLPs were obtained after centrifugation at 30,000 rpm and 4°C for 1.5 h.
Western blot
For Western blot, cells (1 × 106 cells/well) were cultured on a six-well plate at 27°C for 12 h and infected with the recombinant baculovirus rBDV-GP5-M. The cells were collected at 48 h postinfection (hpi) and processed using Radioimmunoprecipitation assay lysis buffer (Beyotime, China). The M and GP5 proteins of PRRSV were detected using mouse anti-Flag (M protein) (1:1,000; Sigma) or rabbit anti-GP5 antibody (1:1,000, kindly provided by Dr. Zhanchang Xie from Jilin Agricultural University) as primary antibodies and then reacted with HRP-labeled Goat Anti-Mouse IgG (H+L) or HRP-labeled Goat Anti-rabbit IgG (H+L) (1:5,000; Beyotime). The results were visualized using the enhanced chemiluminescence (ECL) plus kit (Thermo). The experiments were repeated at least thrice.
Indirect immunofluorescence assay
Sf9 cells (5 × 105 cells/well) were cultured on a 12-well plate at 27°C for 12 h, infected with the recombinant baculovirus rBDV-GP5-M for 48 h. The Indirect immunofluorescence assay was performed as described by Liu et al. (16).
Briefly, cells in 12-well plates were fixed in 4% paraformaldehyde for 10 min at room temperature, washed thrice with PBS for 5 min each, and then treated with 0.25% Triton X-100 for 10 min. The cells were washed thrice with PBS for 5 min each and blocked with 1% Bovine Serum Albumin (BSA) for 1 h. Thereafter, the cells were incubated with 100 μL of mouse anti-Flag (M protein) (1:1,000; Sigma) or rabbit anti-GP5 antibody (1:1,000) for 2 h at room temperature. Then, the cells were washed thrice with PBS for 5 min each and incubated with 100 μL of fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody (1:2,000; Beyotime) or Cy3-conjugated goat anti-rabbit antibody (1:2,000; Beyotime) at room temperature for 1 h. After an additional five PBS-T washes, the cells were incubated with DAPI (Beyotime) at room temperature for 8 min, followed by washing with PBS thrice. Finally, the cells were examined using an Eclipse TE2000-V (Nikon). The experiments were repeated at least thrice.
Transmission electron microscopy
To examine the VLPs using transmission electron microscopy, the purified VLPs were absorbed onto a grid, negatively stained with 1% Phosphotungstic Acid, and examined using a JEM 1200 EX II transmission electron microscope (JEOL, Peabody, MA).
Immunization of mice
Purified PRRSV VLPs measuring 0.5 mL were emulsified with 0.25 mL 2% Poly (vinyl alcohol) (Sigma), 0.25 mL 2% Sucrose solution, 2% Magnesium hydroxide solution, and 4% A5 adjuvant (Sigma); the resulting solution was designated as PRRSV VLP+A5.
BALB/c mice (female, 4 weeks old) were purchased from the Experimental Animal Center of the PLA Military Academy of Medical Sciences (Beijing, China). Mice (n = 30) were randomly divided into three groups for intranasal (i.n.) instillation. Group 1 was immunized with 100 μL of PBS, group 2 was immunized with 100 μg PRRSV VLP, and group 3 was immunized with 100 μg PRRSV VLP+A5. Mice from each group were boosted 3 weeks after the prime immunization using the same inocula used for priming. Immunized mice of each group were divided into two parts. One part (n = 5) was used to analyze T cell immune response, and the other part (n = 5) was used to measure humoral immune responses induced by the VLP. All mice were observed thrice daily for changes in physical appearance and deaths (if any) for up to 6 weeks postimmunization.
Generally, mice were weighed every day to determine the average weight change of the group and were observed for clinical signs of distress. The blood was collected weekly. Six weeks postimmunization, mice were deeply anesthetized by carbon dioxide (CO2) and euthanized by cervical dislocation, and samples were collected.
Collection of serum and lung lavage samples
Blood samples were collected from the tail vein of mice at indicated times and stored at 37°C for 2 h, followed by centrifugation at 4°C, 2,500 rpm for 10 min, and the sera were recovered and stored at −70°C. Sera collected from the tail vein of preimmune mice were used as a negative control. The lung lavage samples were collected, centrifugated at 1,000 rpm, 4°C for 10 min, and stored in aliquots at −80°C.
Detection of specific antibodies
An Enzyme-Linked Immunosorbent Assay (ELISA) Detection Kit (Dakewe, China) was used to detect PRRSV-specific antibodies.
Briefly, 96-well plates were coated with 100 μL of 0.1 μg/mL PRRSV VLPs or polypeptide (BP-2; YQYIYNLTICEL) overnight at 4°C, followed by washing with phosphate-buffered saline-Tween 20 (PBST) thrice and blocking with 100 μL of 1% BSA at 37°C for 2 h. After three washes with PBST, 50 μL of serum samples (1:100) or PBS (negative control) were added to the wells, and the plates were incubated at 37°C for 1 h in a humid environment. After washing with PBST four times, 100 μL of HRP-conjugated goat anti-mouse IgG, IgG1, and IgG2a (1:1,000; Sigma) was added and incubated for 1 h at 37°C. After washing again, 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB) was added into each well in a dark room for 15 min. Finally, 50 μL of 2 M H2SO4 was added to each well, and the absorbance values (optical density, OD) of each well were read at 450 nm using a microplate reader.
Three replicates were used for each sample. The sample was pooled in the group for the analysis.
Lymphocyte proliferation assay
The splenocytes were isolated from immunized mice at 6 weeks post initial immunization, according to a previously described protocol (15).
Briefly, splenic lymphocytes were isolated by centrifugation using a mouse lymphocyte isolation solution according to the manufacturer's instruction (Hao Yang Biological Manufacture Co., Ltd., Tian Jin, China) and resuspended in Roswell Park Memorial Institute (RPMI) 1640 (Hyclone, Beijing, China) supplemented with 10% FBS and 1% penicillin–streptomycin. Then, 50 μL splenocytes (4 × 106 cells/mL) were plated into 96-well plates and cocultured with 50 μL peptide pools (10 μg/mL) (TP-1: CAFAAFVCFVIR; TP-2: RGRIHRWKSPIVVEK; BP-1: DSSTYQYIYNLT; BP-2; YQYIYNLTICEL), which was described previously (7,27,33), at 37°C and 5% CO2 for 3–5 days.
TP-1 and TP-2 are T cell-specific stimulators, and BP-1 and BP-2 are B cell-specific stimulators. Splenocytes cultured with medium alone RPMI 1640 (no stimulation) served as a negative control. Thereafter, 10 μL of WST-1 solution (Beyotime) was added to each well and incubated for 0.5–4 h at 37°C and 5% CO2, and the absorbance values (OD values) of each well were read at 470 nm using an ELISA reader. The results of the proliferation assays were reported as the stimulation index (SI), defined as the ratio between the mean absorbance value from stimulators and the mean absorbance value of the negative control.
Flow cytometry analysis
Hundred microliters splenic lymphocytes (2 × 107 cells/mL) were mixed with 900 μL PBS, centrifuged at 350 g, 4°C for 5 min, and resuspended with 100 μL PBS. Then, cells were stained with 100 μL of FITC anti-mouse CD3 (0.25 μg/106 cells; BioLegend, San Diego, CA), Phycoerythrin (PE) anti-mouse CD4 (1 μg/106 cell; BioLegend), PE anti-mouse CD8a (0.25 μg/106 cells; BioLegend), FITC anti-mouse CD19 (0.25 μg/106 cells; BioLegend), and Antigen presenting cell (APC) anti-mouse CD40 (1 μg/106 cell; BioLegend) at 4°C in the dark for 30 min. After washing twice with 1 mL PBS, the samples were centrifuged at 350 g, 4°C for 5 min and resuspended with 500 μL PBS and analyzed using flow cytometry on a Coulter Epics XL machine (Beckman Coulter).
Statistical analysis
Statistical analysis was conducted using GraphPad 6.0 (GraphPad Software, SanDiego, CA) with a one-way or two-way analysis of variance, followed by the Bonferroni multiple comparison test. The results were statistically significant at p < 0.05. For each separate set of assays, at least three independent experiments were evaluated. The results are expressed as the mean ± standard deviation.
Results
Recombinant bacmid was obtained successfully using the Bac-to-Bac baculovirus expression system
As shown in Figure 1a, GP5 and M genes of PRRSV-1 were amplified and cloned into pMD-19T simple vector to generate plasmids 19T-GP5 and 19T-M. Then, the genes were digested from the plasmids by restriction endonuclease enzymes and subcloned into pFastBac Dual donor plasmid; successively, the resulting plasmid was designated as pFBD-GP5-M (Fig. 1a). The results of digestion with restriction endonuclease enzymes (Kpn I+Xho I and BamH I+Hind III) (Fig. 1b) and sequencing (data not shown) indicated that the donor plasmid pFBD-GP5-M was constructed successfully.
To generate recombinant bacmid, the donor plasmid pFBD-GP5-M was transformed into Competent DH10Bac E. coli cells, followed by the antibiotic selection, resulting in E. coli colonies with recombinant bacmid rBD-GP5-M. As shown in Figure 2, a 1,216-bp of PCR fragment was obtained from the recombinant bacmid using primers pHF and pUC/M13R, indicating that the PRRSV M gene under the control of polyhedrin promoter (PH) was inserted into the bacmid by transposition. Another amplicon, a 2,441-bp fragment, was amplified from the recombinant bacmid using pUC/M13F and p10R, suggesting that the GP5 gene of PRRSV under the control of AcMNPVP10 promoter (P10) was also inserted into the bacmid.
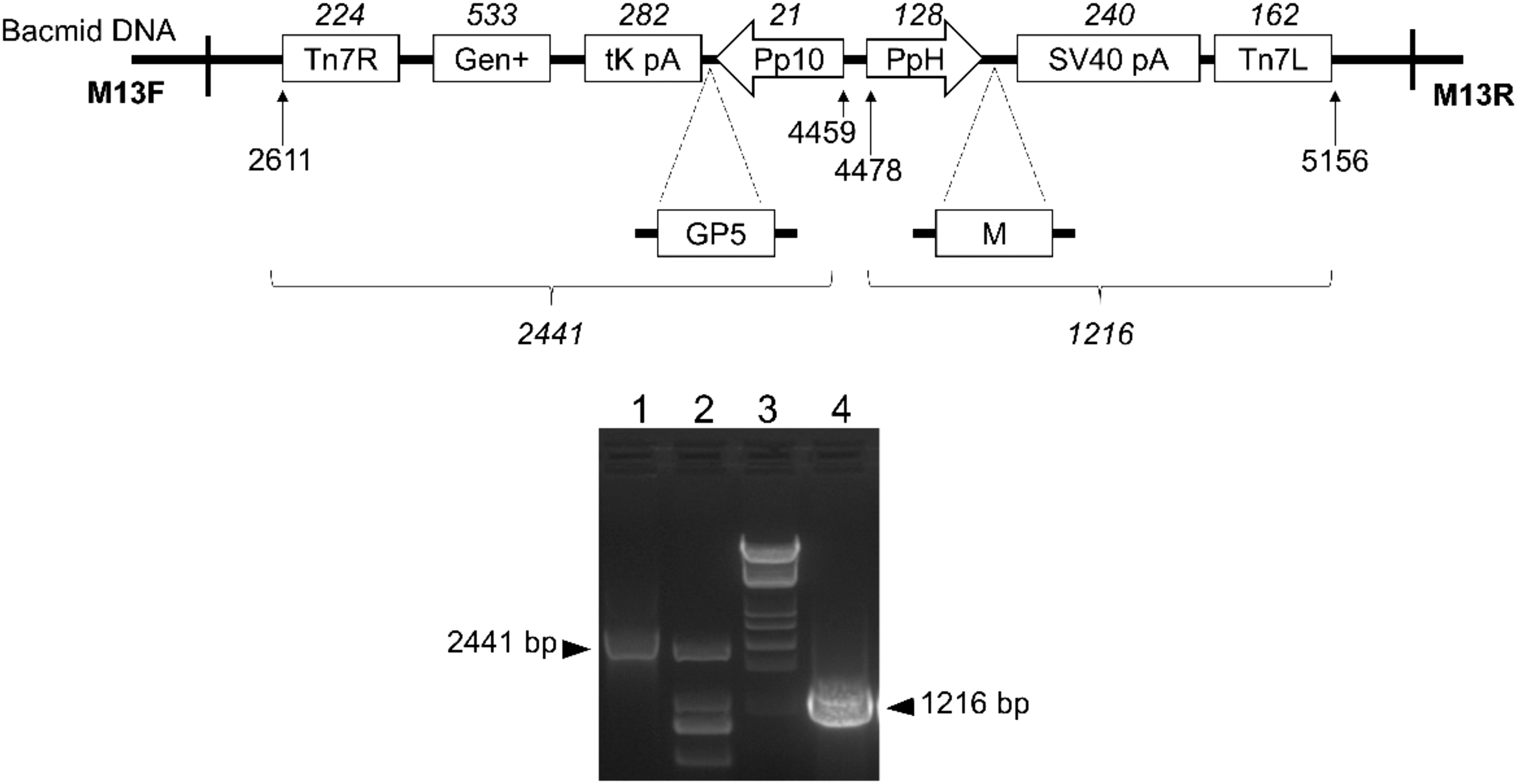
Construction of the recombinant bacmid rBD-GP5-M.
These results demonstrated that recombinant bacmid containing the GP5 and M genes of PRRSV-1 was constructed successfully using the Bac-to-Bac baculovirus expression system.
Recombinant baculovirus expressing exogenous genes was rescued in Sf9 cells using the recombinant bacmid
To obtain recombinant baculovirus, Sf9 cells (106 cells/well) were transfected with the recombinant bacmid rBD-GP5-M. As expected (Supplementary Fig. S1), compared to control cells, the cell diameter was increased and the cells appeared to stop growing at 3 days post-transfection. Subsequently, the cells started detaching and began to lyse at 5 days post-transfection, indicating that the recombinant baculovirus was rescued in Sf9 cells using the recombinant bacmid. Thereafter, the recombinant viruses in the supernatant were purified through centrifugation. The resulting baculovirus was designated as rBDV-GP5-M. Besides, virus titers were evaluated to analyze viral proliferation according to the protocol described by BacPAK Baculovirus Rapid Titer Kit. The virus titer of recombinant baculovirus was up to 2.1 × 108 IFU/mL.
To evaluate the expression of exogenous genes in the recombinant baculovirus, Sf9 cells (5 × 105 cells/well) were infected with recombinant baculovirus rBDV-GP5-M. The results showed that significant fluorescence was observed in the rBDV-GP5-M infected Sf9 cells (Fig. 3a). And a 22-kDa band for GP5 and a 19-kDa band for M protein were detected in rBDV-GP5-M-infected cells (Fig. 3b), while no band was detected in mock-infected cells (negative control). These results indicated that both GP5 and M proteins of PRRSV-1 were effectively expressed in the rBDV-GP5-M-infected Sf9 cells.

Expression of GP5 and M protein in the recombinant baculovirus-infected cells.
To evaluate expression characteristics of GP5 and M proteins, High Five cells (2.5 × 106 viable cells/mL) were infected with rBDV-GP5-M at a MOI of 4. Thereafter, cells and supernatants were harvested at 24, 48, 72, and 96 hpi. As shown in Figure 4, the GP5 protein was detected in cell lysates harvested at 24 to 96 hpi, with the highest expression of the protein at 48 hpi (Fig. 4a). The M protein can be detected in cell lysates at 24 to 72 hpi, and higher expressions were found at 24 and 48 hpi (Fig. 4b). Furthermore, GP5 and M proteins were detected at 48 hpi in the cell supernatant (Fig. 4c, d). These results suggested that the best time to harvest the PRRSV VLPs was at 48 hpi.
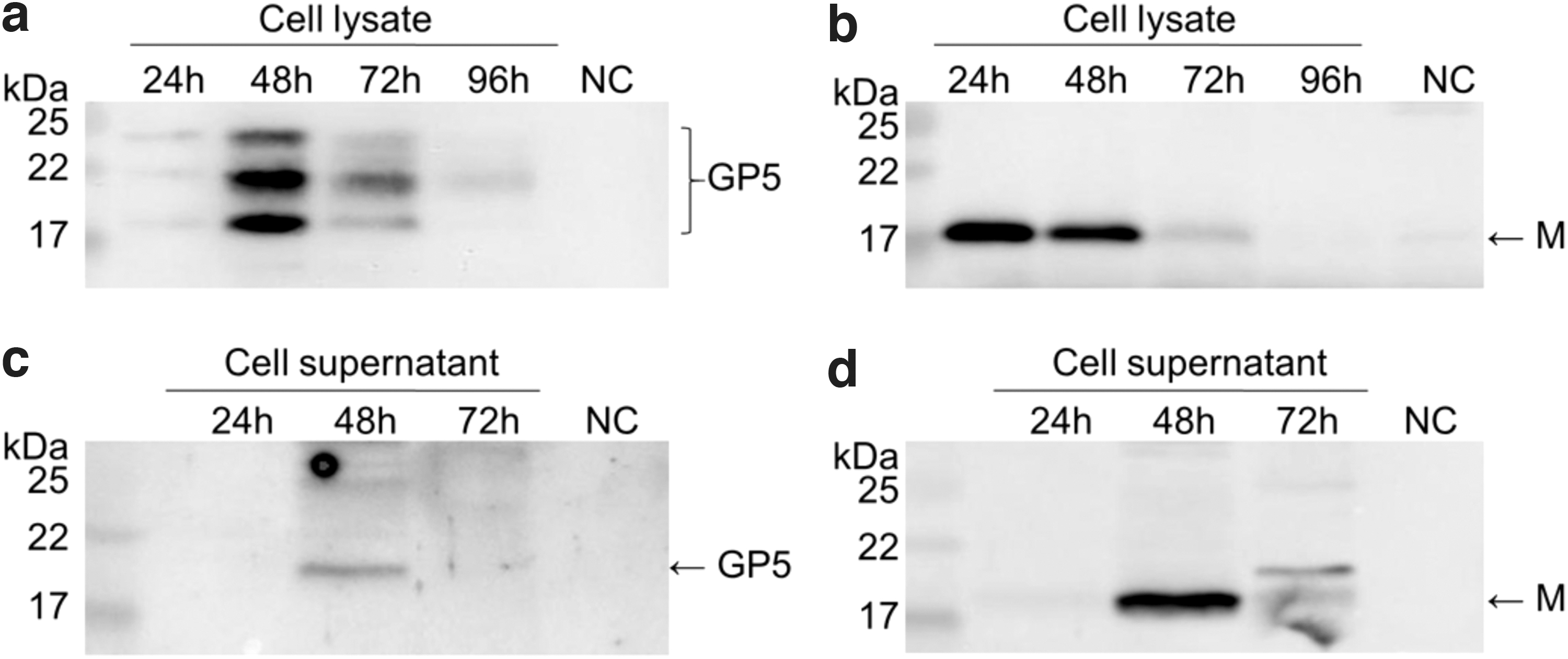
Best harvesting time to produce the PRRSV VLPs. Cells and supernatants were harvested at 24, 48, 72, and 96 hpi through centrifugation. The presence of VLPs in cushion was analyzed by western blot using anti-Flag (M protein) or anti-GP5 antibodies as the primary antibody and HRP-labeled Goat Anti-Mouse IgG (H+L) as the secondary antibody. NC, negative control.
Thereafter, High Five cells were infected with rBDV-GP5-M, and the VLPs were purified from the cell culture media at 48 hpi according to the scheme shown in Figure 5a. As shown in Figure 5b, the GP5 and M proteins were detected in the ultracentrifuged fraction collected from densities between 40% and 60%, which are similar to density values of PRRSV virions. The VLPs showed spherical shapes as typical PRRSV virions (Fig. 5c) and had a peak in size distribution at 50–100 nm (Fig. 5d), about 38.4% of PRRSV VLPs from 90 particles, indicating that the recombinant PRRSV VLPs were generated.

Purification and identification of PRRSV VLPs.
PRRSV VLPs induced effective humoral immune responses and cellular immune responses
Mice were immunized with PBS, PRRSV VLP, or PRRSV VLP+A5 using prime-boost strategy for 6 weeks to evaluate humoral immune responses induced by PRRSV VLPs (Fig. 6a). As shown in Figure 6b, the bodyweight of the mice gradually increased, but there was no significant difference between each group, suggesting that the PRRSV VLP or PRRSV VLP+A5 does not affect the growth of mice. During immunization, total IgG (Fig. 6c) and specific antibody levels (Fig. 6d, e) in sera of immunized groups were increased in a time-dependent manner after priming and boosting with PRRSV VLP or PRRSV VLP+A5, while the antibody level in the PBS group did not increase significantly.
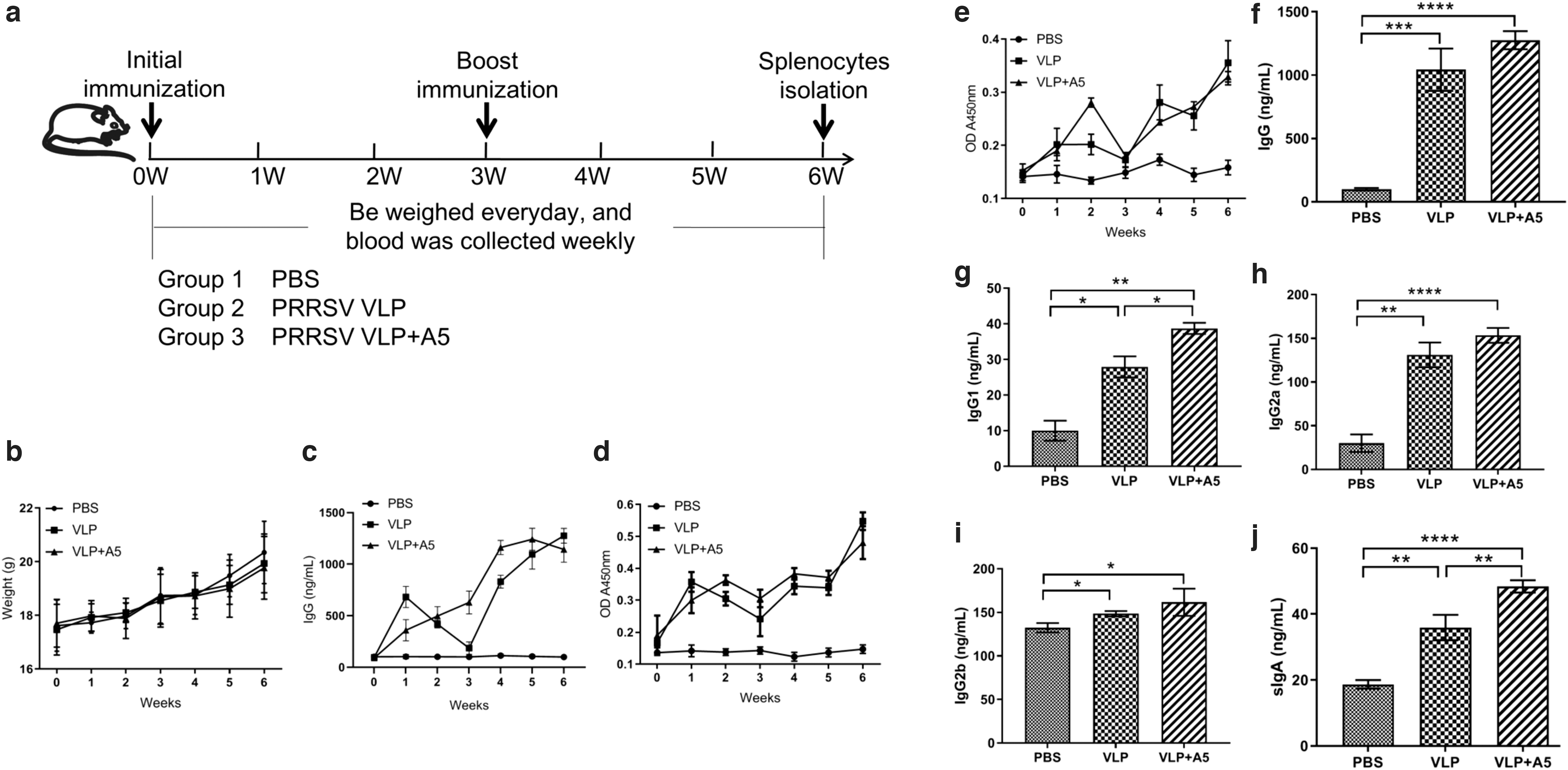
Evaluation of humoral immune responses in different immunized groups. Mice were primed with PBS, PRRSV VLP, or PRRSV VLP+A5 and boosted 3 weeks after the prime immunization using the same inocula used for priming. Blood samples were collected from the tail vein of mice at indicated times after the initial vaccination and used to analyze humoral immune responses using an Antibody Detection Kit. Sera collected from the tail vein of preimmune mice were used as a negative control. The significance of the differences was calculated using the one-way ANOVA (two-tailed, confidence intervals 95%), as indicated by the p-value. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. The results are expressed as the mean ± SD.
Furthermore, PRRSV-specific IgG, IgG1, IgG2a, and IgG2b antibodies in the serum were measured using ELISA at 6 weeks postimmunization. As shown in Figure 6f–i, both PRRSV VLP and PRRSV VLP+A5 induced significantly higher levels of PRRSV-specific antibodies compared with that of the PBS group. Levels of IgG and IgG2a in the PRRSV VLP+A5 group were 13 times higher, and the level of IgG1 was four times higher compared with the PBS group. The IgG and IgG2a levels in the PRRSV VLP group were 10 times higher, and IgG1 levels were 2 times higher compared with the PBS group. Moreover, PRRSV VLP+A5 induced significantly high levels of IgG1 compared with that of PRRSV VLP (Fig. 6g), while there was no significant difference in PRRSV-specific IgG, IgG2a, and IgG2b triggered by PRRSV VLP and PRRSV VLP+A5.
Besides, levels of total sIgA in lung lavage fluid collected from each group at 6 weeks post initial immunization were measured using Mouse sIgA ELISA (Biovision). As expected, the sIgA levels in lung lavage fluid of the VLP-immunized groups were significantly higher compared with that of the PBS group (Fig. 6j). The level of sIgA induced by the PRRSV VLP combined with A5 was significantly higher than that immunized with PRRSV VLP alone. These results indicate that the PRRSV VLP and PRRSV VLP+A5 could efficiently trigger PRRSV-1-specific humoral immune responses in vivo through intranasal immunization. The combination of PRRSV VLP and A5 adjuvant can improve the levels of humoral immune responses.
To evaluate the cellular immune response, the proportion of functional T cell subsets (CD3+CD4+ and CD3+CD8+ T cells) and B cell subsets (CD19+CD40+) in each immunized group was measured. As shown in Figure 7a and b, no significant differences in T cell subset levels were observed between the VLP groups and the control group (Fig. 7a), while the proportions of CD19+CD40+ B cells were significantly higher in the PRRSV VLP and PRRSV VLP+A5 groups compared with that of the control group (Fig. 7b), indicating that both PRRSV VLP and PRRSV VLP+A5 can effectively activate B lymphocyte response.
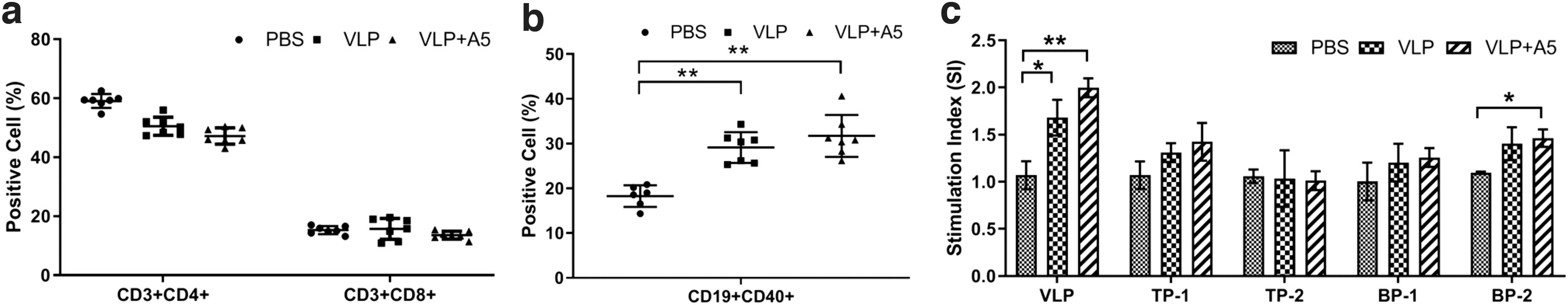
Analysis of cellular immune responses in different immunized groups. The splenocytes were collected from immunized mice at 6 weeks post initial immunization, followed by analysis using flow cytometry or lymphoproliferation assay. *p < 0.05; **p < 0.01.
Furthermore, the proliferation of splenic T lymphocytes from PBS- or VLP-immunized mice was performed by stimulating the cells with purified PRRSV VLP, T cell-specific stimulators (TP-1 and TP-2), or B cell-specific stimulators (BP-1 and BP-2) and determined by WST-1 cell proliferation assay. The results showed that stimulation of lymphocytes with purified PRRSV VLPs resulted in a significant increase in the SI values for splenocytes in the PRRSV VLP and PRRSV VLP+A5 groups compared with that of the control group (Fig. 7c). When the splenocytes were stimulated by T cell-specific stimulators (TP-1 and TP-2) or B cell-specific stimulator BP-1, no significant difference in SI values was observed between the VLP immunized groups and the PBS-immunized group, whereas B cell-specific stimulator BP-2 can significantly enhance the SI values in the PRRSV VLP+A5 group compared with that of the control group (Fig. 7c).
These results indicate that both PRRSV VLP and PRRSV VLP+A5 could enhance specific B lymphocyte proliferation during stimulation by PRRSV VLPs. Taken together, these results demonstrate that the PRRSV VLP and PRRSV VLP+A5 could efficiently stimulate specific B cellular immune responses.
Discussion
The Bac-to-Bac baculovirus expression system is widely used for eukaryotic gene expression in insect cells (14,28). The system provides a rapid and highly efficacious method to generate recombinant baculovirus by site-specific transposition in insect cells. It is recommended to use Sf9 insect cell as the host for baculovirus transfer vector and recombinant baculovirus rescuing, while High Five insect cell is suitable for expressing exogenous genes with high efficiency, resulting in higher protein yields (28,37).
In this study, we inserted GP5 and M genes of PRRSV-1 into the recombinant bacmid, under the control of AcMNPVP10 promoter (P10) and polyhedrin promoter (PH), respectively. Therefore, the recombinant bacmid rBD-GP5-M allows efficient high-level expression of both GP5 and M proteins simultaneously in the baculovirus-infected Sf9 and High Five cells in a nonfusion manner.
To produce VLPs targeting PRRSV-1, both GP5 and M proteins were ultracentrifuged and collected from densities between 40% and 60%, and the diameters of the VLPs were about 50–80 nm. However, unexpectedly, although the VLPs expressing PRRSV1 GP5 and M protein were obtained, the purity of the VLPs is not very high. In addition to the VLPs (accounting for 38.4% of the total protein), it may also contain other components, such as PRRSV GP5 protein or M protein alone or baculovirus (200–300 nm, 17.4%) or baculovirus-related components. Among them, GP5 or M protein has the same immunogenicity as VLP, which can also stimulate a specific immune response.
Notably, both GP5 and M proteins can be detected in cell lysates and supernatants, and expression levels are the highest at 48 hpi, which indicates that the efficiency of protein expression in this system is high.
During last decades, live attenuated and inactivated vaccines of PRRSV are used in swine farms to control PRRSV infection (23). However, their protective efficacy against PRRSV field strains is still controversial. Especially, live attenuated viruses can be recombined with wild-type strain to generate virulent strains, resulting in safety issues (9). On the contrary, VLP-based vaccines are safe and efficient against virus infection (17,21,24).
Furthermore, due to its special morphological structure, VLPs are easily recognized by APC cells and can be presented to activate CD4+ T cells and CD8+ T cells through MHC II and MHC I, thus inducing a cellular immune response (10,11,21,25). Meanwhile, it can interact with Ig receptors of B cells to activate B cells and induce humoral immune responses (10,11,21,25). Moreover, baculovirus and its components can be used as effective immune adjuvants and play important roles in improving immune response (31).
Therefore, we evaluated the humoral and cellular immune responses stimulated by PRRSV VLP and PRRSV VLP+A5 in mice. As expected, the PRRSV VLP and PRRSV VLP+A5 could efficiently trigger PRRSV-1-specific humoral immune responses and specific B cellular immune responses through intranasal immunization.
In addition, to improve the efficacy of the VLP vaccine, we used A5 as an adjuvant in this study. As shown, the combination of PRRSV VLP and A5 adjuvant can improve the level of the immune response. It was reported that purified fraction of Albizia julibrissin saponins (AJSAF) and the recombinant B subunit of the E. coli heat-labile enterotoxin rLTB are promising intranasal adjuvant candidates for the PRRSV vaccine (32,40). Therefore, we will compare the immunological adjuvant activities of A5, AJSAF, and rLTB for PRRSV VLP in the following studies. Besides, studies of the immunogenicity (including neutralization activity) and protective efficacy of the VLPs against PRRSV-1 in animal models, as well as the evaluation of the VLP-based vaccine in swine, are ongoing.
In conclusion, we constructed a recombinant baculovirus rBDV-GP5-M, stably expressing PRRSV GP5 and M protein in this study, which can be used to generate PRRSV VLPs. The PRRSV VLP and PRRSV VLP+A5 could efficiently trigger PRRSV-1-specific humoral immune responses and specific B cellular immune responses through intranasal immunization. The combination of PRRSV VLP and A5 adjuvant can improve the level of the immune response. The PRRSV-1 VLPs generated in this study have greater potential for vaccine development to control PRRSV-1.
Ethics Approval and Consent to Participate
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the Military Veterinary Institute, Academy of Military Medical Science (10ZDGG007). All animal experiments were performed following relevant guidelines and regulations.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Footnotes
Authors' Contributions
Conceptualization, RLZ and LC; methodology, DSW and XW; formal analysis, LTY and WSP; writing-original draft preparation, RLZ and LC; supervision, RLZ and JNY; and funding acquisition, RLZ and LC. All authors critically read and contributed to the article, approving its final version.
Author Disclosure Statement
The authors declare that they have no competing interests. All authors have approved the final version of the article.
Funding Information
This work was financially supported by the Research Unit of Key Technologies for Prevention and Control of Virus Zoonoses, Chinese Academy of Medical Sciences [Grant No.: 2019RU059], the National Natural Science Foundation of China [Grant No.: 31772747, 31702210], the Jilin Province Science and Technology Development Projects [Grant No.: 20200402043NC]. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.