
Abstract
Host exposure to pathogens engage multiple pathogen recognition receptors (PRRs) including toll-like receptors (TLRs); recruit intracellular signaling adaptor proteins primarily myeloid differentiation primary response protein 88 (MyD88) for activating downstream signaling cascades, which culminate in the production of type I interferons (IFNs), proinflammatory cytokines, and chemokines; and impede pathogen replication and dissemination. However, recent studies highlight that absence of MyD88 increased antiviral type I IFN induction, and MyD88−/− mice showed a higher survival rate compared with the low survival rate of the MyD88+/+ mice, implicating MyD88 limits antiviral type I IFN response. As a single infectious agent may harbor multiple PRR agonists, which trigger different sets of TLR-initiated immune signaling, we examined whether MyD88 inhibition during stimulation of cells with more than one TLR–ligand would augment type I IFN. We stimulated human U87- and TLR3-transfected HEK293–TLR7 cells with TLR–ligands, such as lipopolysaccharides (LPS) (TLR4–ligand) plus poly I:C (TLR3–ligand) or imiquimod (R837, TLR7–ligand) plus poly I:C, in the presence of compound 4210, a previously reported MyD88 inhibitor, and measured IFN-β response using an enzyme-linked immunosorbent assay. Our results showed that when U87- or TLR3-transfected HEK293–TLR7 cells were stimulated with TLR–ligands, such as poly I:C plus LPS or poly I:C plus R837, IFN-β production was significantly increased with MyD88 inhibition in a dose-dependent manner. Collectively, these results indicate that during more than one TLR–ligand-induced immune signaling event, impairment of antiviral type I IFN response was restored by inhibition of MyD88 through MyD88-independent pathway of type I IFN signaling, thus, offer a MyD88-targeted approach for type I IFN induction.
Introduction
The pathogen recognition receptors (PRRs), including toll-like receptors (TLRs), the retinoic acid inducible gene I (RIG-I)-like receptors (RLRs)/melanoma differentiation-associated gene-5 (MDA-5), are the basic features of the innate immune system, sense pathogens, or pathogen-associated molecular patterns (PAMPs) (5,17,32,40,46). Most of the pathogens are detected by more than one class of these innate immune receptors (5,17,32,40,46). The engagement of these receptors with PAMPs then initiates intracellular signaling cascades through the recruitment of intracellular signaling adaptor proteins such as myeloid differentiation protein MyD88, MyD88 adaptor like (Mal), also known as toll-interleukin receptor (TIR) domain containing adaptor protein or (TIRAP), TIR-domain containing adaptor protein inducing interferon-β (IFN-β) (TRIF), TRIF-related adaptor molecule, and sterile-a-and armadillo motif-containing protein, (7,10,17,19,35,49,52,53), which ultimately cause the activation of transcription factors and trigger diverse innate signaling pathways. Activation of these TLRs/RLRs-initiated intracellular adaptor-mediated proinflammatory signaling cascades in infected cells ensures a robust innate immune response through the expression of type I interferons (IFNs) and proinflammatory cytokines. However, a given innate immune signaling pathway, essential against some pathogens, may be counterproductive for responses against other immune signaling pathways (3,15,22,27,36,46,50), and antagonism or interference between the individual pathways, eventually may shape the immune response (33). The underlying mechanism of how such a signaling interference or co-operation operates with exposure to pathogens and the specific host signaling component(s) involved in shaping the antiviral innate immune response particularly type I IFN response increasingly appears to be revealing (20,33).
Activation of immediate innate immune response, in particular to virus infection, primarily relies on the synthesis of type I IFN, secreted by various cell types that act as the first, and most rapid, line of host defense (6,45). In particular to TLR pathway of inducing type 1 IFN response, host cellular factor particularly signaling adaptor proteins play a critical role. The ligand binding to TLRs activates signaling by two main pathways: one through MyD88 is used by all TLRs except TLR3 and is shared by IL-1 and IL-18 receptors, and the other through TRIF, solely engaged by TLR3 and TLR4. However, besides TLR3 and TLR4, phagosome localized TLR2/CD36 was also described to elicit TRIF-mediated pathways of inflammatory cytokine response (9). The MyD88 pathway induces early activation of nuclear factor κB (NF-κB) and activation of the activator protein 1 (AP-1) transcription factors. TRIF mediates the TLR3 and TLR4 signaling pathways and is distinguished by the activation of interferon regulatory factor 3 (IRF3) and IRF7, as well as a late NF-κB response and consequential expression of type I IFN (11,12). Notably, the context of TLR signaling of IFN-α/β induction, which modulates the development of both the innate and adaptive immune systems, has gained much attention (30,47), and antiviral immunity in the context of TLR signaling is becoming clearer with regard to the role of TLR adaptors, namely MyD88 and MyD88-adaptor-like (MAL/TIRAP), in the negative regulation of alternative TLRs (24,28,42,43). For example, although MyD88 activates all TLRs except TLR3, it also functions as a negative regulator of TLR3. Thus, it appears that MyD88 plays a considerable regulatory role in the adaptation of host innate immunity (42). In addition to TLR-IL-1Rs, MyD88 is also engaged in the signaling event when stimulated with IFN-γ or MHC class II ligands (16,29,31).
In vitro studies suggest that RIG-I and MDA-5 receptors both detect RNA viruses, and poly I:C, and have emerged as key sensors of viral RNA, which activate IRF-3 and IRF-7, resulting in the induction of the antiviral IFN-β. However, during viral infections, MyD88 has been shown to be significantly upregulated. For example, infection with Coxsackie virus B or venezuelan equine encephalitis virus (VEEV) infection leads to significant upregulation of MyD88 (13,41), which exerts an inhibitory effect due to its ability to sequester IRF3 away from TLR3 or TRIF-mediated signaling. In this regard, MyD88 impairs IFN-β induction that is vital to clear the infection (42,43). Furthermore, it has been shown that IFN-β gene induction was significantly enhanced in MyD88 and Mal/TIRAP-deficient cells after stimulation with poly I:C and after treatment of cells with Mal-inhibitory peptide (38,39,42,43). MyD88−/− mice showed a dramatic higher survival rate (86%) in contrast to a low survival rate (35%) in the MyD88+/+ mice after CVB3 infection (13). Previous results from our laboratory and others described that stimulation of cells with poly I:C in the presence of a MyD88 or Mal inhibitor, augmented IFN-β (38,42). Recent publications on innate immune regulation indicate that distinct combinations of signaling pathways are activated in response to a given pathogen (20). Although recruitment and activation of the adaptor proteins (MyD88 or TRIF) are critical for the induction of cellular innate immunity, dual activation of MyD88 and TRIF has been detected after virus infection (37), and often evokes an opposite effect on inflammatory gene expression (44). MyD88 upregulation has been reported to impair antiviral IFN-β and Regulated on Activation, Normal T cell Expressed and Secreted (RANTES) production. It is also recognized that a single infectious agent harbors multiple PRR agonists and triggers the activation of different sets of receptors mediated through signaling adaptor proteins in mounting innate immune response (25,34). However, it is yet unknown whether inhibition of MyD88 during stimulation of human cells with more than one TLRs–ligand, similar to pathogen-like exposure, would augment IFN-β induction through MyD88-independent signaling pathway. We report in this study that human cells stimulated with more than one TLR–ligand, in the presence of a MyD88 inhibitor, compound 4210, resulted in an increased IFN-β response through the TLR3-dependent pathway.
Materials and Methods
Reagents
Primary anti-MyD88 antibody was obtained from AnaSpec, Inc., (San Jose, CA) and Alexis Bio-chemicals (San Diego, CA). Anti-β-actin antibody was purchased from Cell Signaling Technology (Danvers, MA). Human IFN-β (h IFN-β) ELISA kit was purchased from PBL Assay Science (Piscataway, NJ). Human glioblastoma cell line (U87), which constitutively expresses TLR3 and TLR4, was used to examine IFN-β response and dual TLR–ligand stimulation was obtained from the Cell Culture and Hybridoma Core (USAMRIID). Poly I:C (high molecular weight) and lipopolysaccharides (LPS) were purchased from Sigma-Aldrich (St. Louis, MO). HEK293–TLR3 and HEK293–TLR7-transfected stable cell lines were obtained from Invivogen. MyD88 inhibitor compound 4210 was described elsewhere (1). The structure and the detailed synthesis of the MyD88 inhibitor, compound 4210, used for this study were described elsewhere (1,8).
Stimulation of human cells with dual TLR–ligands and IFN-β assay
U87 cells (1 × 106 cells/well) were cultured with Escherichia coli LPS (1 μg), and poly I:C (10 μg), or HEK293–TLR7 blue cells (1 × 106 cells/well) were cultured with R837 and poly I:C (10 μg) with or without compound 4210 (100 to 3.7 μM). After 20 h, cell culture supernatant was collected for measuring h IFN-β. We detected IFN-β in cell culture supernatant as early as 4 h of stimulation using different cells with TLR–ligands. However, we detected higher levels of IFN-β after 20 h of stimulation (38). We also detected IFN-β in cell culture supernatant after 6 h of VEEV infection treated with MyD88 inhibitor (38). Therefore, for consistency, we measured IFN-β after 20 h of stimulation in the culture supernatant. h IFN-β ELISA kit (VeriKine™ Human IFN-β ELISA Kit; PBL InterferonSource, Piscataway, NJ) was utilized to measure IFN-β by a sandwich immunoassay technique, according to the manufacturer's protocol. In brief, to the precoated wells containing polyclonal antibody to h IFN-β, h IFN-β standards and culture supernatants (100 μL) were added, and incubated for 1 h. The cells were then washed 3 × . The enzyme (HRP)-labeled anti-h IFN-β monoclonal antibody (100 μL) was then added to form an antibody–antigen complex, where the h IFN-β is sandwiched between two layers of antibodies (primary and the enzyme-labeled secondary antibody), and incubated for 1 h, at room temperature. Temperature colorimetric substrate, a colorimetric substrate (100 μL), was used to activate the enzyme reaction. Stop solution was added to each well after 15 min incubation and the plate absorbance was read using the Infinite M200 Pro, TECAN spectrophotometer at 450 nm. Quantitation of IFN-β was done from a standard curve (0–400 pg) of h IFN-β.
Cell culture and transfections
One day before transfection, human HEK293–TLR7 cells (1 × 106 cells/mL) were cultured in six-well plates containing Dulbecco's Modified Eagles's medium, and supplemented with 2.5% fetal bovine serum (Invitrogen, Carlsbad, CA), sodium pyruvate (1 mM),
Results and Discussion
Double-stranded RNA (dsRNA) and intermediate byproducts that are synthesized during the replication of many viruses are recognized by more than one PRR, including TLRs and RLRs/MDA-5, and thus are involved in IFN-α/β gene induction. We previously reported that TLR3-dependent induction of IFN-β was inhibited with over expression of MyD88 in HEK293–TLR3 cells (38). Our study also showed that MyD88− deficiency increased the production of IFN-β with poly I:C stimulation and inhibition of MyD88 increased the phosphorylation of IRF3 that is linked to IFN-β induction [(38), and Supplementary Figure S1]. Earlier reports also described that in Listeria monocytogenes infection, TLR-triggered MyD88 signaling pathways suppress type I IFN gene induction (33). The reports suggest that because innate immune cells express various types of these innate immune receptors, distinct combinations of signaling pathways activated in response to a given pathogen, which may co-operate or interfere with one another in a way that might be beneficial or antagonistic to shape the host immunity, and in this process, intracellular signaling adaptor protein MyD88 plays a predominant role. Consistent with these reports, Hotz et al. (20) reported that TLR and RLR signaling are reprogrammed in opposite directions after virus detection. In the event of multiple TLR-triggered innate immune induction, recruitment and activation of the host signaling adaptor proteins MyD88 or TRIF are essential for MyD88-dependent and MyD88-independent TRIF-mediated signaling pathways for the induction of type IFN response. It has also been reported that dual activation of MyD88 and TRIF has been exhibited after a virus infection (37) and often provokes an opposite response on inflammatory gene expression (44). Our earlier report demonstrated that overexpression of MyD88 in the HEK293–TLR3 cell line and HEK293–MyD88-deficient cells, transfected with plasmid puno-TLR3 and stimulated with poly I:C (dsRNA), reduced IFN-β production compared with nontransfected HEK293 cells (Supplementary Fig. S1). This upregulation of MyD88, as could occur with many viral infections, has been described to affect the antiviral type I IFN response. To substantiate this notion that MyD88 upregulation with multiple TLR–ligand engagement interferes with antiviral type I IFN, such as induction of IFN-β, in this study we used U87, a human glioblastoma cell line (U87), which constitutively expresses TLR3 and TLR4. U87 cells after stimulation with more than one TLR–ligand, such as LPS and poly I:C alone, or in combination in the presence or absence of a MyD88 inhibitor, compound 4210, we measured IFN-β in cell culture supernatant. Our results showed that when U87 cells were stimulated with TLR–ligands, such as poly I:C in combination with LPS, IFN-β production was slightly decreased. In contrast, IFN-β production was significantly (six- to sevenfolds) increased in a dose-dependent manner when stimulated in the presence of compound 4210 (Fig. 1 and Supplementary Table S1). These results suggest that TLR-triggered activation of signaling events is not always beneficial for the host, sometimes restrict in an antagonistic way and impair antiviral innate immune response, such as type I IFN, which functions as a first line of defense particularly against viral pathogens.
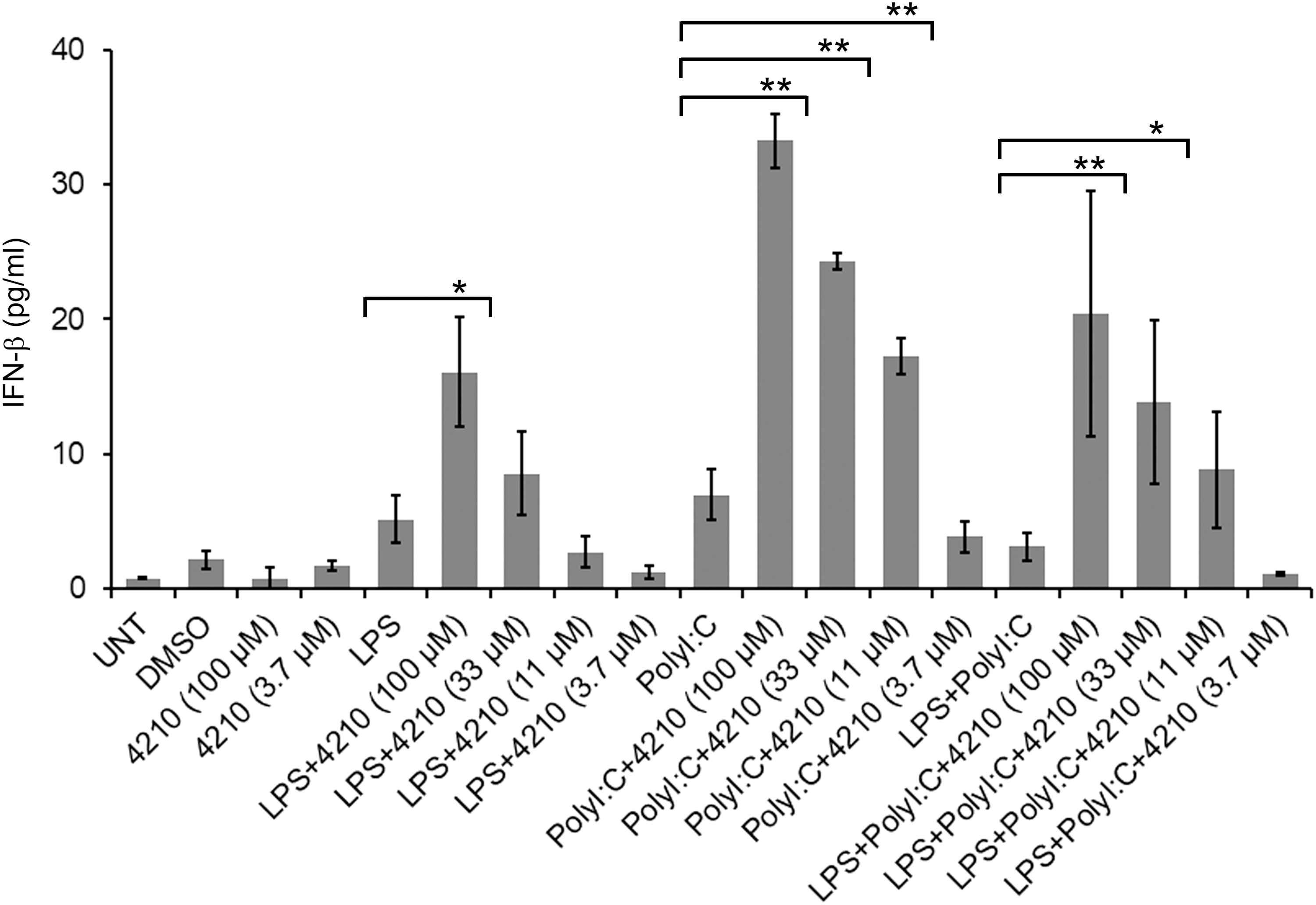
Stimulation of U87 cells with LPS and poly I:C in the presence of a MyD88 inhibitor increased the production of IFN-β. U87 cells (1 × 106) were cultured for 20 h with either Escherichia coli LPS (1 μg), or poly I:C (10 μg), or in a combination of LPS and poly I:C, respectively, with or without varying concentrations of compound 4210 (100 to 3.7 μM). The cell culture supernatants were collected and measured for IFN-β by enzyme-linked immunosorbent assay (ELISA)as described in the Materials and Methods section. Cells treated with DMSO alone were used as control using equivalent volume that contained to highest 100 μM concentration of compound 4210. Also compound 4210 alone at 100 and 3.7 μM concentration in DMSO was also used as controls. Data are presented as an average (pg/mL) from multiple experiments after stimulation of cells; asterisk indicates a significant difference between the pairwise comparisons of compound 4210 treatment and without compound 4210 treatment. LPS stimulation of cells IFN-β at 100 μM compound 4210 treatment (p ≤ 0.0243), poly I:C stimulation of cells IFN-β at 100, 33, and 11 μM compound 4210 treatment (p ≤ 0.0001), (≤0.0005), (p ≤ 0.0036), respectively. LPS and poly I:C stimulation of IFN-β at 100 and 33 μM compound 4210 treatment (p ≤ 0.0045), and (p ≤ 0.0134), respectively. Data are represented from three independent experiments. DMSO, dimethyl sulfoxide; ELISA, enzyme-linked immunosorbent assay; IFN-β, interferon-β; LPS, lipopolysaccharides.
In general, innate immune cells such as monocytes/macrophages are known to respond to LPS through both the MyD88-dependent and MyD88-independent (TRIF-dependent) pathways to produce an inflammatory response, suggesting the importance of MyD88 in both pathways (43). MyD88 functions as a negative regulator of TLR3 (42), and MyD88 deficiency has also been reported to increase production of IFN-β with poly I:C (dsRNA) stimulation or viral infection through TLR3-TRIF pathways (13,42,43). Consistent with these reports, in this study, our results suggest that upregulation of MyD88 with LPS stimulation implicated TLR3-dependent suppression of IFN-β response. Reports from our laboratory and others suggest that the increase in the type I IFN response, through MyD88 inhibition, occurred simultaneously with increased IRF3 phosphorylation (38,42). To further corroborate the observation, besides LPS, we stimulated cells with more than one TLR–ligand, such as imiquimod (TLR7–ligand) which is known to activate MyD88-mediated signaling, and poly I:C. We used human HEK293–TLR7 cells, transfected with a plasmid containing TLR3 (Puno-TLR3-GFP), then stimulated with imiquimod and poly I:C, and measured IFN-β levels in cell culture supernatant. Our results showed a significant increase (sevenfold) in the production of IFN-β, in the presence of compound 4210 treatment, in a dose–dependent manner, compared with cells in the absence of compound 4210 treatment (Fig. 2 and Supplementary Table S2). Controls such as HEK293–TLR7 cells stimulated only with imiquimod in the presence of compound 4210 also showed an increase in IFN-β induction, notably more with TLR3 transfection without poly I:C stimulation. These results are consistent with our earlier report that inhibition of MyD88 in the presence of compound 4210, in U937 cells or THP1 cells stimulated only with LPS or poly I:C, increased in IFN-β production (38). We also noted an upregulation of MyD88 from a basal level with time in HEK293–TLR3 with poly I:C stimulation (Supplementary Fig. S2). These results suggest that an increase in MyD88, after imiquimod stimulation, negatively affected the MyD88-independent pathway of IFN-β production, because inhibition of MyD88 augmented IFN-β. This is consistent with a previous report that showed that MyD88 signaling has been shown to block IFN-β responses with bacterial infection (33). The results are also consistent with a report that showed that the antiviral immune response to poly I:C requires TLR3, and treatment with a MyD88 peptide inhibitor increased type I IFN and other immune-stimulatory genes in a lower vertebrate, the Japanese Flounder (Paralichthys olivaceus) infected with megalocytivirus (54). Additional recent reports also provide evidence in support of the negative effect of MyD88 on the IFN response and increased survival of IPS-1−/− MyD88−/− mice after infection with severe fever with thrombocytopenia syndrome virus, which suggests that MyD88-independent mechanisms are involved in the survival of IPS-1−/− MyD88−/− mice (51). Although virus proteins are known to impede with the induction of antiviral type I IFN signaling events to evade host antiviral innate immunity, in this context it appeared to be innate immune signaling adaptor protein MyD88 upregulation contributing to the dampening of the innate immune response, which is critical for viral clearance. During VEEV or Coxsackie virus B3 infection, or TLR3-dependent poly I:C stimulation, MyD88 upregulation has been shown to negatively influence host antiviral type I IFN and RANTES production (13,42,43,41). On the contrary, enhanced antiviral IFN-β and RANTES production was observed in MyD88-deficient (MyD88−/−) mice that likely led to significantly increased protection from lethal Coxsackievirus infection (13). Similar reports were also described with Herpes simplex virus type 1 (HSV-1) infection (18), and Nipah virus infection (21). It has also been reported that although the transcription factor IRF7 is essential for HSV-1 infection for the induction of IFN-α/β genes through the virus-activated MyD88-independent pathway and the TLR-activated MyD88-dependent pathway in HSV-1–infected splenic pDCs, however, the later pathway is not essential for the in vivo innate immune response against this virus (18). Consistent with these mentioned reports, more recently Totura et al. reported that TLR3 signaling, through TRIF, contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection (48). During early onset of an infection, the induction of a strong type I IFN system is indispensable for vertebrates to control viral infections as they modulate immediate-concerted innate immune responses in a balanced manner that promotes antigen presentation, and also natural killer cell functions (23).
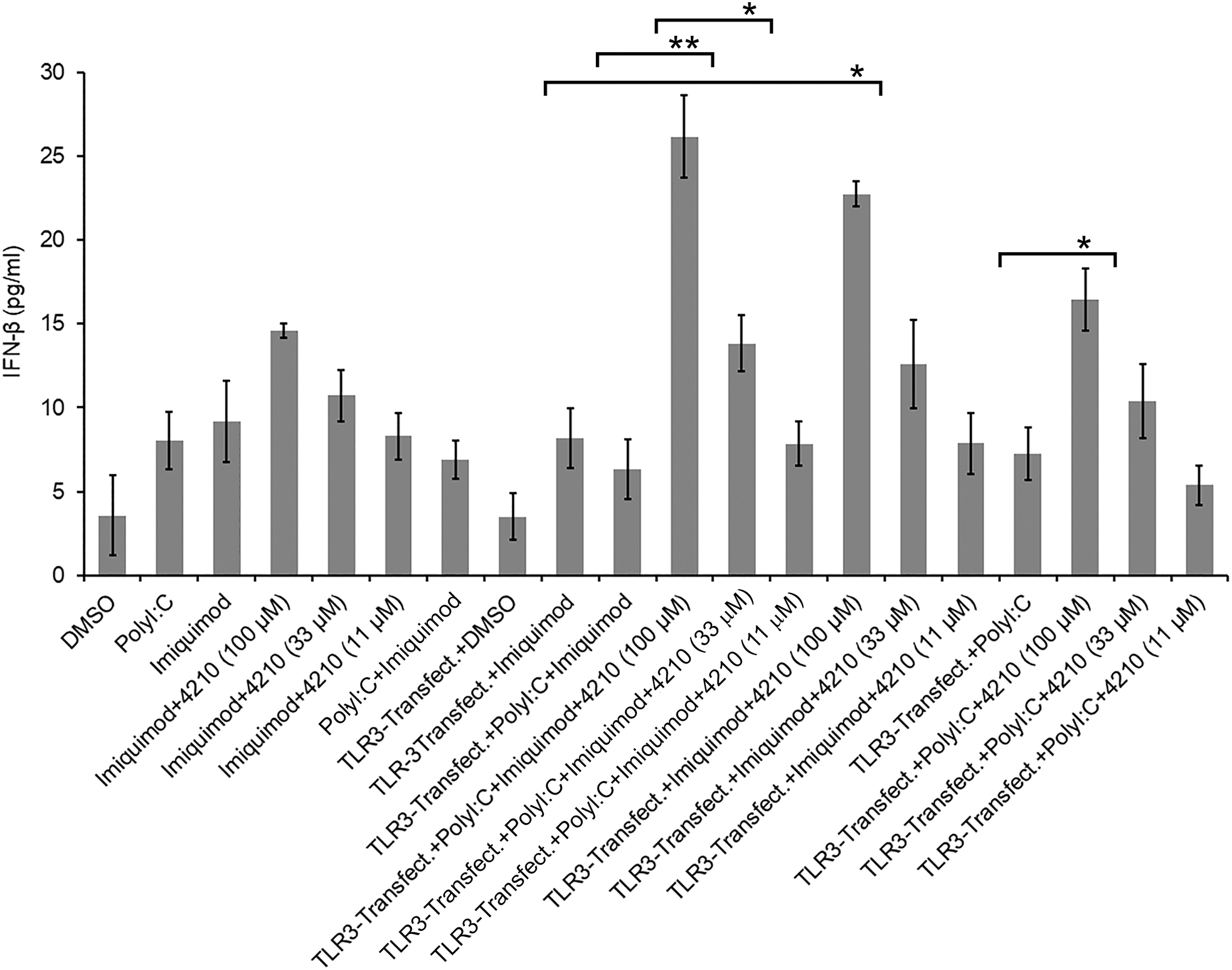
HEK293–TLR7 cells expressing TLR3 stimulated with imiquimod and poly I:C in the presence of a MyD88 inhibitor increased IFN-β production. HEK293–TLR7 cells (1 × 106/mL) were treated with imiquimod, or HEK293–TLR3 cells (1 × 106/mL) were transfected with plasmid Puno-TLR3-GFP using Lipofectamine 2000 (Invitrogen) method according to the manufacturer's instructions as described elsewhere (37) and treated with poly I:C, and poly I:C plus imiquimod without or with varying concentration of compound 4210 (100 to 3.7 μM). Cell culture supernatants were collected at 20 h postinfection for measuring IFN-β by ELISA (PBL Interferon Source's VeriKine). Cells treated with DMSO alone were used as control using equivalent volume that contained to highest 100 μM concentration of compound 4210. Data are presented as an average (pg/mL) from multiple experiments after stimulation of cells; asterisk indicates a significant difference between the pairwise comparisons of compound 4210 treatment and without compound 4210 treatment. TLR3–ligand (imiquimod) stimulation of transfected HEK293–TLR7 cells expressing TLR3, treated with poly I:C in the presence of compound 4210 at 100 μM concentration, increased IFN-β (p ≤ 0.0033). Similarly, HEK293–TLR7 cells stimulated with imiquimod in the presence of compound 4210 increased IFN-β (p ≤ 0.0036). HEK293–TLR7 cells expressing TLR3, treated with both poly I:C and imiquimod in the presence of compound 4210, increased IFN-β at 100 and 33 μM concentration (p ≤ 0.0011), (p ≤ 0.0233), respectively, at 100 and 33 μM compound 4210 treatment (p ≤ 0.0045) and (p ≤ 0.0134), respectively. TLR, toll-like receptor.
Type I IFN is an integral component of first line of defense for the host antiviral innate immune system, which has been used for treating infectious diseases (4,14,26). Our previous results indicated that a small molecule inhibitor of MyD88 augmented IFN-β production after poly I:C stimulation of multiple cell types (38). The increase in the type I IFN response, with MyD88 inhibition, was concurrent with increase in phosphorylation of IRF-3, which is indicative of TRIF-IRF-Axis–mediated IFN-β induction. MyD88 upregulation linked with many viral infections and MyD88 interaction with IRFs both impair the antiviral type I IFN response that has been described previously (13,38,42,43). As MyD88 deficiency has been reported to increase the type I IFN response (2,13,18), it is suggested that upregulation of My D88 negatively regulates type I IFN response. In this study, our results suggest that stimulation of multiple classes of innate immune receptors, such as TLRs in a cell, leading to activate distinct signaling pathways, mediated by host signaling adaptor protein, sometimes interferes and suppresses type I IFN gene induction that is critical for antiviral immunity. Therefore, a therapeutic approach targeting MyD88, a host factor, involved in the impairment of antiviral innate immune response, appeared to be helpful for inducing type I IFN against certain viral pathogens.
Our results, in conjunction with earlier reports, suggest that MyD88 exhibits a characteristic of a novel pharmacologic target for the induction of host antiviral IFN response. Induction of innate immunity and its regulation during viral infections are precisely a combination of host factor(s) that likely contribute in perpetuating innate signaling cascades and, thus, set a balance of those signals that may cooperate or interfere with one another in ways that can be beneficial or antagonistic to affect the magnitude, shape, and composition of host immunity (20,33). Consistent with previous reports, our data provide underlying mechanisms that in the context of multiple TLR–ligands initiated activation of innate immune signaling cascades, particularly the induction of antiviral IFN response could be augmented with MyD88 inhibition in viral infection, and thus may be utilized for a potential therapeutic approach for host-directed antiviral therapy.
Footnotes
Authors' Contributions
K.U.S. outlined, designed, and supervised the study, compiled data, drafted the article text with another author contributing to the writing process. C.M.R. performed cell-based infection assays, analyzed data, and reviewed the article.
Disclaimer
Views expressed in this article are those of the authors and do not purport to reflect official policy of the U.S. Government or USAMRIID/ARL administrators. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the U.S. army.
Acknowledgments
We gratefully acknowledge the contribution of our longtime research collaborator, Prof. Julius Rebek, Jr., and his group (The Scripps Research Institute) for synthesizing compound 4210, utilized in this study, and the technical help of Shahabuddin Alam in measuring cytokines and performing other assays. We also acknowledge the contribution of David Fretter for statistical analysis and Lorraine Farinick for preparing the figures.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was partly supported by The Defense Threat Reduction Agency grant originally awarded to K.U.S. (Grant No. CBM.THROX.03.10.RD.006).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.