
Abstract
Till today, the globe is still struggling with the newly emerging infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and known as coronavirus disease 2019 (COVID-19). It has resulted in multiple fatalities from SARSs all around the world. A year after the global pandemic, the World Health Organization (WHO) has reported more than 79 million confirmed cases of COVID-19 and over 1.7 million deaths, making it one of the worst and most difficult pandemics encompassed in the modern history. The ongoing triad of escalating infections, mortality, and economic loss has urgently called for recognizing SARS-CoV-2 cell entry mechanisms as a crucial step in the initial stages of infection and to which possible interventional strategies should be targeted. To mediate host cell infections, Coronaviruses utilize the immunogenic studded spikes glycoproteins on its surface as a key factor for attachment, fusion, and entrance to host cells. Herein, we shed the light on a potential strategy involving disruption of SARS-CoV-2 S protein interaction with host cell receptors through design of neutralizing antibodies targeting receptor binding domain in S1 subunit, small peptide inhibitors, peptide fusion inhibitors against S2, host cell angiotensin converting enzymes 2 (ACE2), and protease inhibitors, aiming to pave the way for controlling viral cell entrance. In this review, we also highlight the recent research advances in the antiviral drugs that target the highly exposed spike protein, aiming to stem the COVID-19 pandemic.
Introduction
The last two decades have witnessed an upsurge of viral outbreaks, especially of the deadly coronaviruses (CoVs). The CoV outbreaks have started back in 2003 with severe acute respiratory syndrome coronavirus (SARS-CoV) in Asia followed by 2012 middle east respiratory syndrome coronavirus (MERS-CoV) that was first identified in Saudi Arabia and then sporadic outbreaks among other countries of the Arabian Peninsula occurred. Currently, from late 2019 till now, a novel coronavirus that was subsequently named SARS-CoV-2 had emerged in Wuhan City, China's Hubei Province (79). Coronavirus disease 2019 (COVID-19) had rampantly run across the globe, leading the World Health Organization (WHO) to entitle the outbreak as a pandemic by March 11, 2020 (96). Globally as reported by WHO, there have been around 92,506,811 confirmed cases of COVID-19, including 2,001,773 deaths, as of January 16, 2021 (96).
Taxonomically, CoVs belong to the order Nidovirales, suborder Cornidovirineae, family Coronaviridae, and two subfamilies, namely, Letovirinae and Orthocoronavirinae. The former subfamily includes the Alphaletovirus genus, while the latter based on the genome structure is classified into four genera: Alpha, Beta, Gamma, and Delta CoVs that contain 17, 12, 2, and 7 unique species, respectively (73,39). CoVs have zoonotic reservoirs that can likely cross the species barriers, and some have extended to human. So far, seven human COVs (HCoVs) that mainly belong to either Alpha (229E and NL63) or Beta genera (OC43, HKU1, MERS-CoV, SARS-CoV, and SARS-CoV-2) were identified. Epidemiological studies had revealed that bats are the natural reservoir of the three novel Beta-HCoV outbreaks, and Arabian camels (4), masked Himalayan civet cats (30), and other unknown reservoirs (22) are the potential intermediate hosts for MERS-CoV, SARS-CoV, and SARS-CoV-2, respectively. Notably, our understanding and significant identification of such HCoV reservoirs will intensely aid in predicting the place and time of possible epidemics.
Virus Structure and Genomic Organization
CoVs are enveloped viruses, with a nonsegmented, positive-sense RNA strand resembling the largest genome size (26–32 Kb) among the other studied RNA viruses (82). Generally, the genomic organization of such CoVs follows a characteristic order: At 5′ end a open reading frame 1a/b (ORF1a/b) representing about 67% of the whole genome translated into two polyproteins (pp1a/pp1b) and codes for 16 nonstructural proteins (NSPs 1–16) with exception of Gamma-CoVs that lack NSP1. NSPs 1–16 play specific roles in viral replication, including RNA dependent-RNA polymerase and virion assembly, but still the functions of NSPs 2 and 11 remain not fully understood. At 3′ end other ORFs mainly code for important structural proteins as spike (S), envelope (E), membrane glycoproteins (M), nucleocapsid proteins (N), and hemagglutinin esterase (HE) among some Beta-CoVs, in addition to other accessory proteins interspaced within structural proteins that are unique in number, function, and sequence to SARS-CoV-2. Commonly, structural proteins along with accessory proteins are essential to regulate structure and function of virion assembly and viral pathogenesis (17).
The genomic sequence of SARS-CoV-2 consists of 10 ORFs; about two-thirds of viral RNA are present in the first ORF coding for pp1a/pp1b, NSPs 1–16 and the remaining ORF codes for the structural proteins S, M, E, and N and accessory proteins (33). In comparison to SARS, recent mutations in NSP 2 and NSP 3 through the presence of glutamine, serine, and proline at 501, 723, and 1,010 positions along the sequence of SARS-CoV-2 could affect its properties. The stabilizing mutation encoded by glutamine that falls in the endosome-associated-protein-like domain of the nsp2 could possibly provide increased stability to protein and explains the increase contiguously of SARS-CoV-2. In contrast, the destabilizing mutation near the phosphatase domain of the nsp3 proteins encoded by serine and proline is expected to create a steric effect suggesting a potential unique mechanism for SARS-CoV-2 (2,76).
Although CoV genome can function directly as efficient mRNAs in cells due to the 5′methylguanosine cap structure along with poly (A) tails, still only a portion of replicase genes that precedes structural and accessory genes are translated. Then, the replicase genes will use template genomic RNA to produce complementary negative strands where mRNA encoding viral proteins are transcribed (46). As a result of discontinuous transcription, a nested set of subgenomic RNAs at 3′ end is synthesized characterizing Nidovirales order that stands for nested in Latin.
SARS-CoV-2 Spikes Glycoproteins
Among all of the structural proteins, the most important is the S glycoproteins that project from the spherical virion surface giving them the appearance of royal crown as depicted under electron microscope, prompting the name CoVs as shown in Figure 1 (85). On the virion surface, the S protein with size of 180–200 kDa assembles into clove shape trimers consisting of extracellular N terminus, transmembrane, and short intracellular C terminal domains (11). With a total length of 1,273 amino acids in S protein, the extracellular ectodomain is composed of two critical subunits: S1 subunit (14–685 residue) that contains an N-terminal (S1-NTD) and a C-terminal (S1-CTD) subdomains, where both of which can function as a receptor-binding entity and S2 subunit (686–1,273). The former subunit is responsible for recognition and binding to host receptors through the receptor binding domain (RBD) (60) and the latter for fusion between viral envelope and host cell membrane through putative hydrophobic fusion peptide (FP) and two heptad repeat donated as HR1 and HR2 (86). The FP and HR elements are followed by a transmembrane domain and a C-terminal intracytoplasmic tail that plays a role in S protein sorting. In addition to the important role of spike proteins in regulating viral entry into the host cells, they are considered highly antigenic determinants that are usually targeted by host antibody response. Such diversity of S proteins among CoVs had enhanced their interaction and response to different receptors. It had been reported that dipeptidyl peptidase 4 (also known as CD26) (59) and angiotensin converting enzymes 2 (ACE-2) receptors are functional receptors for MERS related CoVs and SARS-CoV (56), including SARS-CoV-2 (111), respectively. Except for the mouse hepatitis CoVs, the majority of other CoVs, as well as SARS-CoV-2, utilize S1-CTD to bind to their receptors. In comparison with SARS-CoV RBD, more atomic interactions through the engaged residues and the larger buried surface areas had reflected the high binding affinity of SARS-CoV-2-CTD spike protein to ACE2 receptors (97). Moreover, the conformational transition of SARS-CoV-2 S glycoprotein between its closed and open states as based on down and up positions of the RBD, the S protein can be in a receptor inaccessible closed or accessible open state, respectively (31). In CoV infections, the step of receptor binding along with membrane fusion is considered the initial important steps that should be targeted for inhibiting viral entry. Different targeted therapeutic strategies against SARS-CoV-2 cell entrance mechanism that involves targeting: the two pathways of virus entry, virus receptors on host cells, spike proteins, and virus fusion to host cells will be reviewed in the coming part.
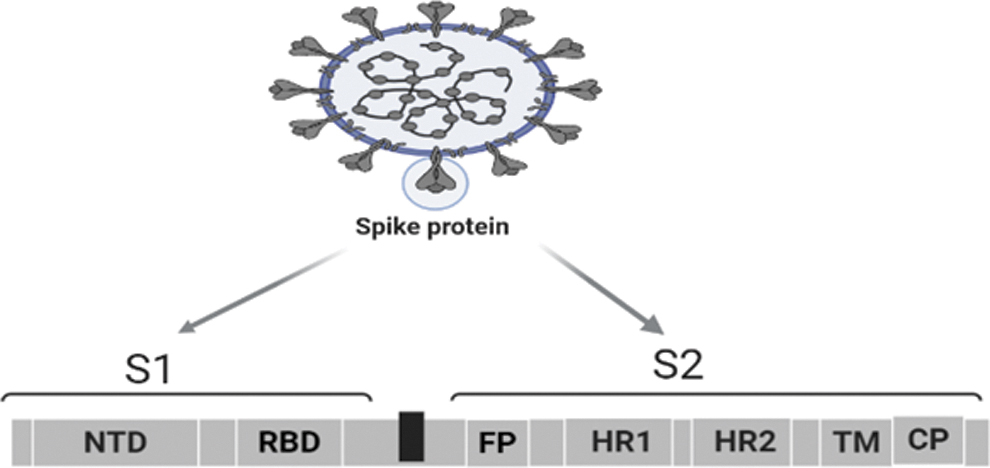
Schematic presentation of SARS-CoV-2 spike domain (created with
The genome of one strain of SARS-CoV-2 isolated from a patient with COVID-19 in Egypt has been sequenced and submitted into the National Center of Biotechnology Information (NCBI) GenBank (MW010256). The tertiary structure model of the spike glycoprotein (protein_id = QNT08785.1) of this Egyptian strain was carried out using Swiss-Model software (
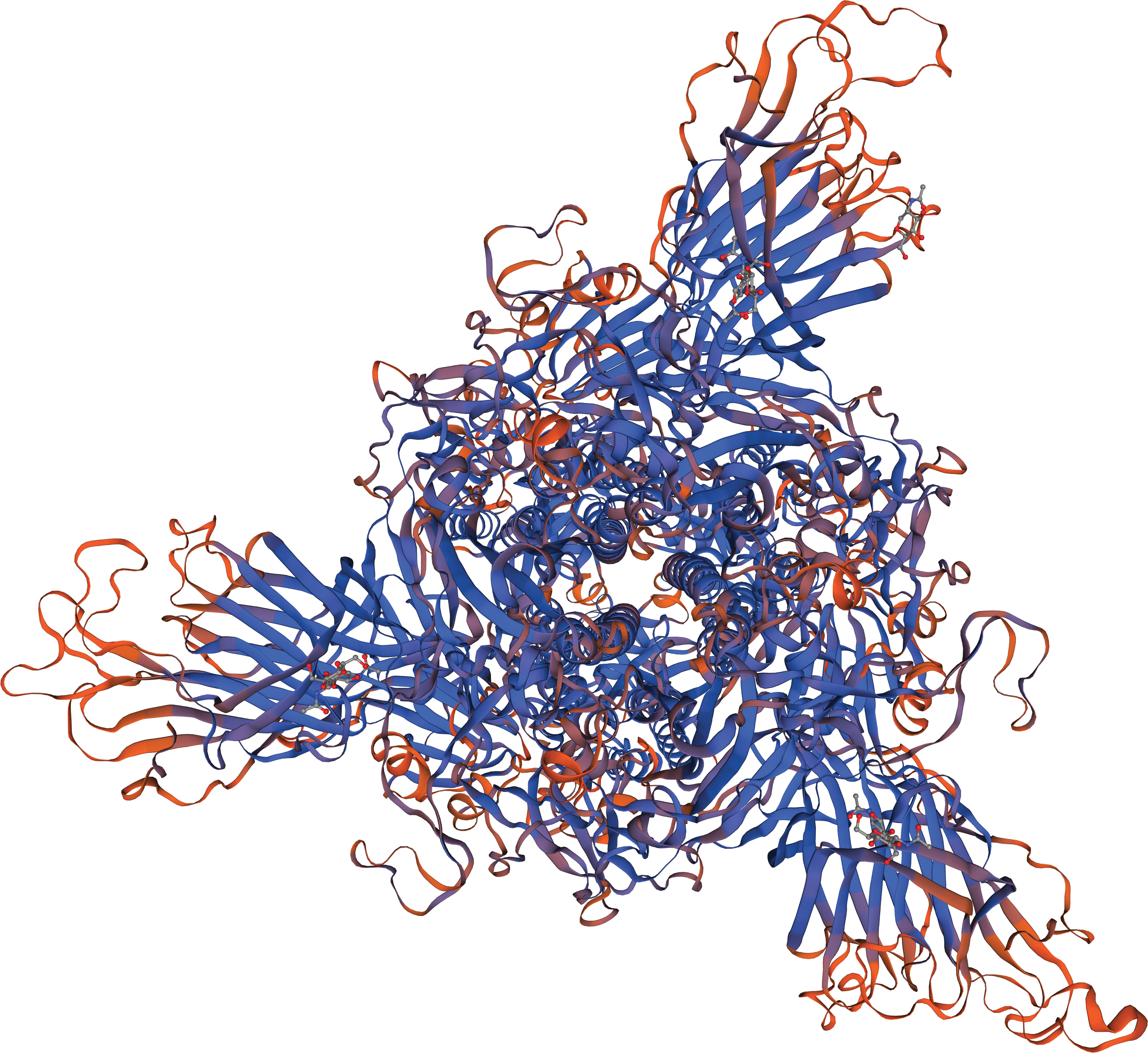
The putative tertiary structure of spike glycoprotein (protein ID = QNT08785.1) of SARS-CoV-2/human/EGY/hCoV-19-Egypt-MASRI-14-4-2020/2020 isolate using Swiss-Model software.
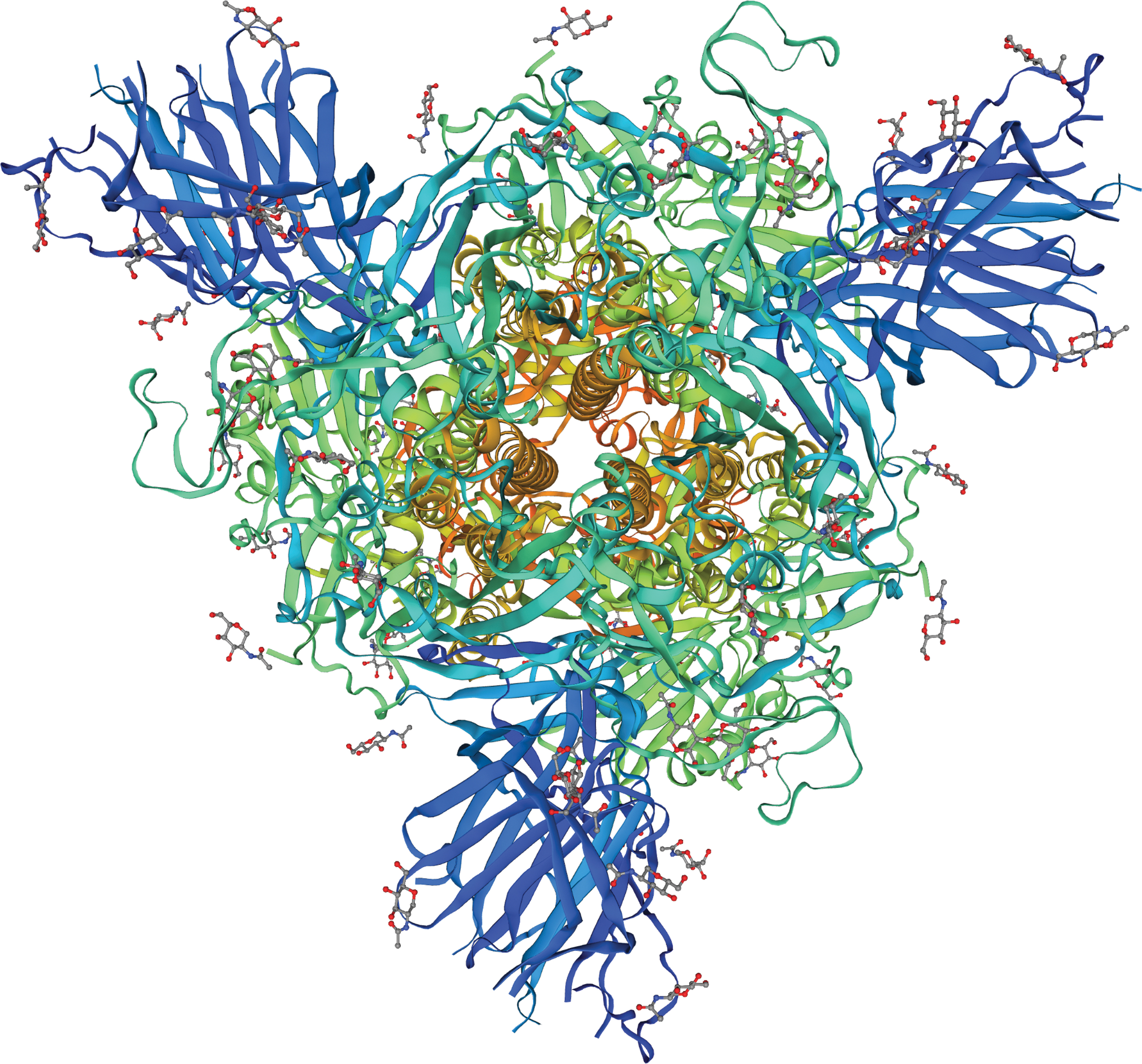
The tertiary structure of spike glycoprotein template (protein template = 7cwm.1) (109) using Swiss-Model software.
Targeting Viral Entry
Although the engagement of ACE2 host cell receptors are important for viral entry of SARS-CoVs, the subsequent entrance steps can vary and are specific to cell type. Generally, the endocytic and/or the cell surface nonendosomal pathways mediate entrance of CoVs to the host cells. The endosomal pathway is facilitated by acidic pH (4.5) and the pH dependent endosomal cysteine protease cathepsins, while other host proteases such as transmembrane protease serine 2 (TMPRSS2) and TMPRSS11D play a critical role in nonendosomal (clathrin independent) pathway (116,108). Briefly, in the former pathway binding of S1 domain to ACE2 receptors will trigger conformational changes in S2 domain that result in internalization and subsequent membrane fusion by Cathepsin L during endocytosis. In contrast, the latter pathway is followed by additional cleavage of S proteins into the two domains by host serine proteases namely type-2 transmembrane serine protease (104).
SARS-CoV-2 can use both endosomal and nonendosomal pathways for entry where low pH enhances its activity inside cells. Thus, it is relevant to use drugs that increase pH and impairs activity of lysosomal enzymes as lysosomotropic agents. Phosphoinositide lipids, namely, phosphatidylinositol 3-phosphate 5-kinase (PIKFYVE), regulate a part of endosome formation and are also required by virus to infect human cells through endocytosis. PIKFYVE kinase inhibitors as Apilimod previously known to inhibit production of interleukins (IL-12, IL-23) and YM201636 were found to significantly reduce entry of pseudo-typed SARS-CoV-2 bearing S protein as reported by Ou and colleagues (70). In addition, Apilimod had strongly inhibited infection by authentic SARS-CoV-2 strain with a half maximal inhibitory concentration (IC50) of ∼10 nM (47), and AI therapeutics had recently announced start of Phase II Trial of Apilimod dimesylate: (AI Therapeutics, a Guilford, Conn.-based biopharmaceutical company formed by Yale, unpublished data). Similar to apilimod, YM201636 had also blocked viral entry and infection by other viruses like African swine fever virus or Ebola virus (28).
In addition, a two-pore segment channel that regulates conductance of calcium ions was reported to be important in endocytosis of SARS-CoV-2. Tetrandrine, a traditional Chinese medicine with a recently discovered antagonistic effect against two pores channels, was reported to be effective in vitro against SARS-CoV-2 pseudovirus. Despite limited pharmacological background about tetrandrine, multiple evidences suggest that it could be an effective measure in the containment of the viruses when used in clinical practice (25,35,43).
Endosomes have an acidic interior that is mainly maintained through the activity of vacuolar proton pump known as V-ATPase (67). Hence, several efforts have been made to target this process to block the entry or replication of CoVs. Recent in vitro studies had revealed that endosome acidification inhibitors namely bafilomycin-A1 had inhibited the entry of porcine delta CoV (112) and SARS-CoV-2 at 100 Nm (70). In addition, bafilomycin A1 reversibly inhibits late stage fusion between lysosomes and autophagosomes thus preventing maturation of autophagic vacuoles (105). Similar to other RNA virus, CoVs each in its own unique way can likely utilize the cellular autophagy pathway to enhance its replication (5,64), and therefore, drugs targeting autophagy could show promising results in the search for anticoronavirals. In addition to bafilomycin-A1, other macrolides resembling azithromycin, clarithromycin, erythromycin, and telithromycin have been proposed as valuable options for viral respiratory infections characterized by over-exuberant innate inflammatory response like COVID-19 due to their anti-inflammatory and immune-modulatory effects (66,71). Azithromycin targets granulocytes where it markedly concentrates in lysosomes and particularly affects accumulation, degranulation, and apoptosis of neutrophils (69).
For several decades, chloroquine has been the drug of choice for treatment of malaria owing to its good efficacy, relative safety, and low price. By the addition of a hydroxyl group, a hydroxychloroquine had replaced the semisynthetic chloroquine due to its decreased level of renal (84), ocular toxicity (44), and conserved efficacy. Although the molecular mechanism of action of such antimalarial drugs may not be fully clarified, prior studies have reported the interference of chloroquine and hydroxychloroquine with CoVs through a series of steps (pre- and postinfections). Both drugs are weak bases that are easily trapped in acidic compartments leading to the elevation of lysosomal pH and subsequent reduced capacity of the virus to infect the cell (37,77). In postentry stages, the antiviral activities are related to inhibition of glycosylation of the newly synthesized viral proteins (89). The in vitro anti SARS-CoV-2 activity of hydroxychloroquine was found to be more potent than chloroquine as the 50% maximum effective concentration for the former was 6.14 and 0.72 μM at 24 and 48 h, while for the latter was 23.90 and 5.47 μM at 24 and 48 h, respectively (110). In addition to their antiviral action, they demonstrated an immune-modulating activity that inhibited cytokine storms and thereby enhanced the antiviral efficacy of the drugs in vivo (95). Despite the previously mentioned potentials, no enough data are available to support the routine use of hydroxychloroquine and chloroquine for treatment of COVID-19 due to the possibility of serious side effects.
Targeting S1 Subunit
Being exposed to the surface, S-glycoprotein would be a potential target for developing neutralizing antibodies, viral attachment inhibitors, and effective vaccines (90). Although all antigenic determinants of S protein could provoke production of neutralizing antibodies, still RBD immunogen can elicit up to five times higher affinity antibodies (74). The S1 RBD mediates the critical step for viral entrance to target cells through a main functional motif known as receptor binding motif (RBM) in RBD. Two regions of the RBM, as well as a region outside it, form the interface between the S protein and human ACE2 (hACE2) receptors (58) and maintain structural stability in RBD (13), respectively. Different approaches to identify multiple monoclonal human neutralizing antibodies were reported by many research teams. The first report by Wang et al. identified 47D11 with a cross neutralizing activity targeting conserved core structure of the S1B RBD. However, such binding was far away from the receptor-binding interface, showing limited ability for spike receptor interactions (94). Hence, more experiments are required to evaluate the effect of combination with a potent neutralizing noncompeting antibody that targets RBD. Wu and colleagues isolated four neutralizing monoclonal antibodies from a convalescent patient; two antibodies named as B38 and H4 had blocked the binding of RBD with ACE2 receptors at different sites (98). Thus, they can bind concurrently, resulting in lower dosage, and may even lessen risk of immune escape in clinical practice. Using high-throughput single cell RNA sequencing and variability, diversity, and joining sequencing of B cell receptor repertoires, Cao and colleagues identified 14 potent neutralizing antibodies recovered from 60 convalescent patients. Among them, BD-368-2 was the most potent one exhibiting IC50 of 15 and 1.2 ng/mL against authentic SARS-CoV-2 and pseudo-typed system, respectively. In addition, BD-368-2 had also shown strong prophylactic and therapeutic efficacy in SARS-CoV-2-infected hACE2-transgenic mice by maintaining body weight and reducing viral copies by 3–4 log cycles (15). Based on enzyme linked immunosorbent assay and flow cytometry, Wan et al. had identified 11 neutralizing antibodies to virus entrance into Vero-E6 cell, and eight of them had shown IC50 within 10 nM. The most potent was 414-1 with IC50 of 1.75 nM, and interestingly without any optimization about 300 mg/L was expressed in Chinese hamster ovary cells, offering great potential on the expense of therapeutic development (91). Another target for developing therapeutic antibodies is the NTD that can potentially hinder S conformational changes required for postfusion stage. However, less neutralizing activity has been recorded to that type of antibodies compared with RBD neutralizing antibodies (19).
Although the phylogenetic analysis of the Beta CoV genus reveals that SARS-CoV-2 is more phylogenetically related to SARS-CoV as they share ∼80% sequence similarity, still sequences originating from bat CoV RaTG13 have shared 96.3% similarity with Wuhan SARS-CoV-2 (72). Generally, this high rate of similarity has raised the researchers hope to reuse or design broad spectrum antiviral agents to minimize the current pandemic. However, the multiple sequence alignment of the major antigenic determinants of S1 proteins across the SARS-CoV from bats, human, or civets and SARS-CoV-2 has shared around 73–76% similarities in the RBD and 50–53% in the RBM (38,92). Despite similarities in structure of RBD-ACE2 interface from SARS-CoV-2 and SARS-CoV, as the root mean square deviation equals 0.68 Å over 139 Cα atoms, still several key amino acid variations at three clusters (N-terminal, central, and C-terminal) were reported by Wu and colleagues (99). Thus, blocking of this binding interface represents an encouraging therapeutic strategy, as it could simply impede cellular uptake of SARS-CoV-2 and the intracellular replication. Based on the molecular dynamic stimulation of α1-helix of ACE2 (a peptidase domain that is important for binding SARS-CoV-2-RBD), SBP1 a 23 mer peptide and SBP2 were extracted. The former is likely stable to bind to SARS-CoV-2 RBD with low nano-molar affinity, while the latter peptide was a reduced analog in the middle of SBP1 peptide and not able to bind to RBD (111). Based on the protease domain of ACE2, another group had mainly targeted the α1 helix in addition to α2 helix and the linker of the β3 and β4 sheets that effectively bind RBD of the SARS-CoV-2. They reported that in comparison to inhibitor 1 that used α 1-helix, inhibitors 2–4 that used α 1, 2-helices had supported each other and had retained a bent shape to provide a full cover of RBD surface (34). Hence, considerable molecular knowledge will pave the way to design selective peptide-based antagonists of the hACE2 receptor with enhanced binding affinity that is capable to disrupt large surface of SARS-CoV-2 RBD/ACE2 interactions.
Under the evolution process for viral fitness, the highly glycosylated spikes protein of SARS-CoV-2 had been undergoing mutations, challenging their use as an ideal target for treatment (75). In March 2020, a substitution of adenine (A) to guanine (G), called D614G, was the first spike mutation to be recorded in public databases, crossing the borders and spreading in many locations throughout the world (40). Although this high rate of transmissibility is a key factor for viral fitness, still confirming the role of the mutation in improving fitness is debatable. Some researchers concluded that D614G had improved viral replication in upper respiratory tract, but fortunately had increased neutralization susceptibility, to help in developing better vaccines to hold the pandemic. In addition, the alteration of S proteins was always accompanied by three other mutations through substitution of cytosine to thymine at 241, 3,037, and 14,408 positions leading to mutation in 5′ untranslated region, silent mutation in the nsp3, and RdRp rendered the amino acid Proline to leucine P323L, respectively (50,62). Moreover, Wang and coworkers had reported the presence of hundreds of new mutations on SARS-CoV-2 S protein, mainly located on RBM. With a total of 194 residues, 89 mutations were on the RBD, and 52 out of 89 mutations were on the receptor-RBM. It is worth saying that they predicted that such mutations on the RBD particularly those on RBM (residues 452, 489, 500, 501, and 505) have high probabilities to mutate into significantly more infectious SARS-CoV-2 strains (16,94). Recently, the United Kingdom has witnessed a new variant through substitution of Asparagine to Tyrosine (N501Y) in RBD. Unfortunately, such a mutation had tendency to increase overall interaction between ACE2 and RBD (26,53). Hence, understanding the dynamics of existing mutations may enhance the prediction of future infection tendency and outcome of potential treatment.
With the start of wave two pandemic, an extraordinary need to develop effective and safe vaccines against SARS-CoV-2 will be a high priority to alleviate and prevent future waves of the current pandemic. Using the computational biology, scientists are mapping immunogenic S protein epitopes to select potential antigens for SARS-CoV-2 vaccines. New radial approaches that target SARS-CoV-2 S glycoproteins, including DNA-based vaccine, mRNA-based vaccines, virus-like particle vaccines, and protein-based vaccine, are currently passing different phases of clinical trials to accelerate vaccine development process (23,3). Recently, BNT162b2 (mRNA-based vaccine) developed by Pfizer in a combined phase 2/3 clinical trial has been granted the first emergency use authorization in the United States and till now it is approved in several countries with an efficacy rate of 95%. The second one to be authorized by United States was RNA-1273 developed by Moderna with an efficacy rate of 94% (88). Despite their approval in several countries to limit the pandemic, still many questions regarding their effectiveness against different age groups and the emergence of side effects with longer follow-up remain to be answered.
Targeting the ACE2 Host Cellular Receptors
About 2 decades ago, a type 1 transmembrane glycoprotein known as ACE2 that shares 42% homology with ACE zinc binding catalytic domain was discovered (24). Unlike ACE, ACE2 has only one active domain that acts as monocarboxypeptidase cleaving a single amino-acid namely phenylalanine (52). ACE2 can catalyze cleavage of angiotensin I (1 –10) and angiotensin II (1 –8) into angiotensin 1–9 and angiotensin 1–7, respectively. Angiotensin II can either bind to type 1 receptor or be processed to give angiotensin 1–7. Over-reactivity of former action leads to hypertension, cardiac, and renal remodeling, while latter action will involve binding of angiotensin 1–7 to a G-protein coupled receptor known as mitochondrial assembly receptor (Mas), resulting in vasodilation and antiproliferative action (41). Hence, activating Mas axis could have potential therapeutic perspectives in circulatory and metabolic disorders.
Since the interaction of RBD with ACE2 receptors remains the most fundamental step for SARS-CoV-2 attachment to host cells, targeting the functional host receptor ACE2 will interfere with the virus entrance from the beginning. In addition to being anchored to plasma membrane with the extracellular domain, ACE2 exists in a soluble circulating form. A human recombinant soluble ACE2 (hrsACE2) prepared by genetic modification in Apeiron Biologics known as APN001 was designed to compete for membrane bound ACE2 (1). Monteil et al. in vitro studies had reported the effectiveness of clinical grade hrsACE2 in reducing viral growth (by a factor of 1,000 to 5,000 times) in Vero cells and effectively neutralized infected engineered organs (human blood vessels, kidney organoids) (68). Currently, hrsACE2 has already passed through phase II clinical trials (NCT04335136) as a treatment for patients with COVID-19. Furthermore, another clinical trial undergoing phase I (NCT04375046) is comparing the effectiveness of a genetically engineered ACE2 like enzyme that is derived from Escherichia coli expression system with hrsACE2 in controlling COVID-19 infection and lung injury.
The attachment of the S protein to ACE2 also triggers the activation of a disintegrin and metalloproteinase domain 17 known as ADAM17. In addition, ADAM17 is known as a membrane “sheddase” that can release ectodomain of a variety of membrane-anchored cytokines and ACE2 enzymes into the extracellular medium as biologically active sACE2 form (29). Of note ADAM is also known as tumor necrosis factor (TNF)-α converting enzyme (TACE) that can assist in the shedding regulation of ACE2. Indeed, the intensified upregulation of ADAM17 mediated ACE2 cleavage coupled with release of TNF-α and IL-6R secretions will lead to additional harmful effects, including increasing inflammatory immune response and coagulation cascade (115). Specific inhibitors to ADAM, including GW280264X, and TACE inhibitors (TAPI-0, TAP1-2) were effective in reducing shedding of ACE2 against SARS-CoV at 1 nM, IC50 value of 100 and 200 nM, respectively (32).
Another promising selective small molecule inhibitor against sACE2 is MLN-4760 that binds to active Zinc site and competes with transition state peptide. Studies revealed that it is the most potent drug in inhibiting catalytic activity of ACE2 with an IC50 of around 440 pM (87). Despite the promising results of direct inhibitory compounds against ACE2, still the vital role of ACE2 in maintaining certain physiological functions should be placed into consideration.
Targeting S2 Subunit
As mentioned earlier the S glycoprotein of CoVs is composed of S1 subunit that binds cellular receptor with its RBD and S2 subunit that plays a significant role in membrane fusion and subsequent entrance of virus to cells. Such binding of the virus to specific host receptor will trigger the crucial step “membrane fusion” that can occur directly at plasma membrane or following endocytic pathway (6). At the molecular level, the metastable S protein structurally rearranges from the prefusion to postfusion configurations. In the former configuration, the S protein is primed (proteolytic cleavage) by host cell proteases into two subunits, and upon binding of S1 subunit to the host cell receptor, it dissociates from the S2 subunit. In the latter configuration, membrane fusion initiates with the insertion of the FP into the target cell membrane, and successively, the HR regions fold back onto each other, causing the formation of a thermostable six-helix bundle structure where the membranes are pulled into close contact and ultimately fuse (54,90). Basically to mediate fusion of viral and cellular membrane, S proteins exhibit significant conformational flexibility from closed (prefusion) to open conformations (postfusion) to expose its RBD (103). Accordingly, interference with membrane fusion of S protein through peptide analogs, mAbs, and proteolytic processing inhibitors could be an attractive therapeutic target that will be highlighted in coming sections.
Currently with advanced techniques, the design of peptides and their mimetics had gained a wide interest among pharmaceutical research due to their potential antagonist effect against various pathogens. In terms of time and specificity, nowadays peptides are smaller fragments of proteins with improved half-life that shows affinity to desired target and low toxicity to accumulate inside the body (12,48). Based on sequencing results, SARS-CoV and SARS-CoV-2 showed high similarity in HR1 and HR2 with identity of 92.6% and 100%, respectively (97). Hence, a potential broad spectrum HCoV fusion inhibitor will be more likely directed toward HR2. Moreover, X-ray crystallographic fusion structures of SARS-CoVs and MERS revealed that HR1 peptides in absence of HR2 did not exhibit antiviral effect due to their tendency of aggregation and inability of forming trimer structures. However, numerous peptides derived from the HR2 were able to inhibit enveloped viruses likely through competitive binding to the HR1 sequences (10,57,101). EK1 a modified HR2 peptide derived from fusion core of OC43-CoV (OC43-HR2P) had shown a broad spectrum of inhibition against five HCoVs, namely, MERS, SARS-CoV, HCoV-229E, HCoV-NL63, HCoV-OC43, with IC50 ranging from 0.19 to 0.62 μM in the cell fusion assays. In comparison to OC43-HR2P, the introduction of charged amino acid as glycine or lysine had enhanced the solubility and the antiviral activity of the EK1. In addition, in vivo study had reported on high safety profiles and protective effect of intranasal delivered EKI, confirming its clinical potential for coming outbreak (100,101). Coupled with the start of the pandemic, the team of the previously described study has designed two fusion inhibitors against SARS-CoV-2 named as 2019-nCoV-HR1P and 2019-nCoV-HR2P. As expected, the latter had potently inhibited cell fusion with an IC50 value of 0.18 μM, while the former did not show significant inhibitory effect. In comparison to 2019-nCoV-HR2P, EK1 peptides were also found to effectively inhibit SARS-CoV-2 pseudovirus infection in 293 T cells with IC50 values of 0.98 and 2.38 μM, respectively (102). For further enhancement of EK1 inhibitory effect a lipidation strategy was used where cholesterol (C) and palmitic acid (P) were covalently attached to the C-terminus of EK1 sequence under the help of a flexible polyethylene glycol spacer to construct EK1C and EK1P lipopeptides, respectively. In comparison to EKI, EK1C4 showed more potent anti-SARS-CoV-2 in membrane fusion and pseudovirus infection with about 241 and 149 fold, respectively (102).
In comparison to traditional antivirals, targeting host protease factors may provide therapies with a broader spectrum of activity and decreased risk of developing resistance. However, their design should be coupled with advanced tool of bioinformatics to minimize interfering with host protease that are expressed at different levels with a similar preferred substrate range (81). Indeed, as earlier described the proteolytic priming of S glycoprotein among CoVs involves host proteases as endosomal cathepsins, cell surface TMPRSS proteases, furin, and trypsin. Such cleavage to allow entrance of SARS-CoV and other CoVs occurs at two characteristic sites, one at boundary (S1/S2 boundary) and the other within SAR-CoV S2 domain (S2′) (65). By focusing on cleavage sites, Jaimes et al. reported that former site encompasses a PRRA (P = proline, R = arginine, A = alanine) starting at 681 amino acid sequence among SARS-CoV-2 only, while latter site and FP are firmly conserved among SARS-CoV-2, SARS-CoV, and bat-CoV (SL-CoV-RaTG13). In addition, an R residue at 685 position was detected against SARS-CoV-2, and these three R residues had facilitated proteolytic processing by the pro-protein convertase family of proteases transmembrane serine protease (42). Hence, selective inhibition of host proteases that meddles with virus entrance may act as a valuable antiviral strategy to control viral propagation.
Depending on their role in regulating cell–matrix interactions, a multidomain type II transmembrane serine protease known as TMPRSS2 is mostly expressed in prostate in addition to other cells like colon, kidney, liver, lung, and pancreas. Of note, TMPRSS was only coexpressed in a subset of ACE2 cells (83). In Japan, an irreversible serine protease inhibitor known as camostat mesylate, characterized by aromatic polar guanidine recognition elements, had been used clinically against a wide panel of diseases, including liver fibrosis, pancreatitis, and prostate cancer (36). Although earlier evidence had reported that high concentration of camostat (100 μM) was only effective to partially block entrance of SARS (65%), still the remaining 35% of antiviral activity could be achieved by addition of endosomal cathepsin inhibitors as E64D (49). Moreover, previous studies had reported on in vivo effectiveness of camostat in animal model that was indicated by a 10-fold reduction in SARS-CoV titer in Calu-3 airway epithelial cells (14) and an enhancement of survival rates among 60% of mice (114). Recently, different interventional studies (NCT04608266, phase 3), (NCT04455815, phase2/3) have been developed to evaluate safety and efficacy of camostat mesylate among patients with SARS-CoV-2 infections in ambulatory adult patients that are able to stay at home. Another broad spectrum structure analog to camostat known as nafamostat mesylate (nafamostat) had been used as short acting anticoagulant in disseminated intravascular coagulation and anticancer and anti MERS-CoV S mediated protein fusion as reported by Yamamoto and colleagues (107). Recently, same research group had reported that nafamostat mesylate was 10 fold more active than camostat mesylate in cell fusion assay of SARS-Cov-2. Moreover, nafamostat mesylate showed anti SARS-CoV-2 in Calu-3 cells by targeting TMPRSS2-dependent plasma membrane pathway with an EC50 = 10 nM (106). Recently 2 clinical studies (NCT04418128, phase 2/3 and NCT04352400, phase 2/3) are evaluating the clinical efficacy of nafamostat mesylate for hospitalized COVID-19 patients with pneumonia.
In addition to using nonendosomal pathway, SARS-CoV-2 can also enter cells through endosomal pathway that is mediated by cathepsins B and L (CatB/L) (55). Cysteine cathepsin proteases are involved in many physiological activities and are highly abundant in endosomes and lysosomes to break down proteins and class II major histocompatibility complex. The CatB/L are cysteine endopeptidase that are made up of three characteristic catalytic residues cysteine, histidine, and asparagine at position 25,159, and 175, respectively. It is worth noting that in terms of shape and volume, the active sites of CatB/L did not show significant differences, while their substrate specificity remains different (20,80). Hence, targeting Cat through inhibition will have the advantage of first blocking CoVs on host cell surface and second blocking release of viral material inside the endosome. Inhibitors for CatB/L namely E64D, K11777, and SID26681509 showed enhanced in vitro activity against SARS-CoV-2 activity (45). Ou et al. treated HEK 293/hACE2 cells with a broad inhibitor for Cat B, H, L (E64D) and specific Cat L inhibitor (SID 26681509), and their results revealed 92.5% and more than 76% reduction in SARS-Cov-2 entrance, respectively (70). In addition, at least seven Cat L inhibitors with different pharmacological effect and toxicity have been tested to block CoV infection of human cells. The most potent was K11777 a vinyl-sulfone, and three of its analogs (SMDC256122, SMDC256159, and SMDC256160) had shown the lowest IC50 in pseudotyped SARS-CoV infection inhibition assay (114). Although K11777 good pharmacokinetic parameters in animal models had encouraged the examination of this potential molecule in blocking SARS-CoV-2, still being irreversible Cat L inhibitor had often led to undesirable toxicity. In contrast, a reversible Cat L inhibitor known as oxocarbazate (CID23631927) was effective at low concentration to block SARS-CoV pseudotyped virus entry in human cells (78). This promising anti SARS-CoV activity of oxocarbazate derivatives could be a hopeful aspirant to control current outbreak.
Furin, as well as its related proprotein convertase (PC) family members, and trypsin are also other host proteases that play a role in entry and biogenesis of CoV proteins. Alarmingly, a furin cleavage complex that enhances pathogenicity had been uniquely detected at S1/S2 boundary of SARS-CoV-2 S protein, but not in SARS-CoV or other lineages of β-CoVs (21,27,61).
Bestle and coworkers had reported on high efficiency of the synthetic furin inhibitor (MI-1851) a peptide mimetic against SARS-CoV-2 multiplication in human airway cells (8). Regarding its potent activity to reduce virus titer by up to 75 fold at 10 μM, MI-1851 had been combined with various TMPRSS2 inhibitors to enhance antiSARS-CoV-2 (8). The combination results were promising, and the team reported a more potent activity in comparison to single use of any serine protease inhibitors (8). In addition, another furin-mediated peptide inhibitor for S proteins known as decanoyl-Arg-Val-Lys-Arg-chloromethylketone (CMK) had completely abolished virus cytopathic effects in addition to significant reduction in SARS-CoV titer (7). Interestingly, Cheng and colleagues had tested four furin/PC inhibitors, including CMK and naphthofluorescein (a small molecule inhibitor), in SARS-CoV-2-infected VeroE6 cells. The immune-blot analysis of cell lysates showed a remarkable decline in the processed S protein fragments parallel with reduced N protein. In addition, inhibiting the virus infectivity in VeroE6 at different time points revealed that CMK affects the early stage of the virus replication cycle (entry), while naphthofluorescein had only decreased the viral RNA and protein level (18). Accordingly, the simultaneous use of these two related furin inhibitors would target SARS-CoV-2 replication cycle at different points.
Trypsin, a prototype of serine endopeptidase, is normally expressed in digestive system, kidney, and to a lower level in lung and has shown similar substrate specificities to TMPRSS family. Moreover, its cleavage at a neutral or a slightly basic pH (8.0) had made it able to take precedence over the acidic cathepsin-mediated S cleavage, facilitating direct viral entry to plasma membrane (51,61). A bovine pancreatic trypsin inhibitor known as aprotinin had been approved in Russia in its aerosolized form to alleviate influenza symptoms without recorded side effects (113). Bojkova et al. had reported the effectiveness of aprotinin as anti-SARS-CoV-2 over another trypsin inhibitor (prolastin) in three different cell cultures named as Caco2, Calu-3, and primary bronchial epithelial cell air–liquid interface cultures (9). Despite its effectiveness in preventing virus from entry, aprotinin slows down fibrinolysis process (63) and thus should be used with caution in severe COVID-19 patients suffering from disseminated intravascular coagulation. All the potential targets of SARS-CoV-2 spike glycoprotein are delineated in Figure 4.

Potential targets of SARS-CoV-2 spike glycoprotein (created with
Concluding Remarks
Since its emergence by the end of 2019, the highly contagious SARS-CoV-2 causing COVID-19 had rapidly become a major concern for humanity. To overcome its deadly effect, the scientific community is in a rush to adopt several effective potential anti-SARS-CoV-2 drug targets that will add to the prophylactic and treatment protocols. More attention had been directed toward targeting the spike protein that plays a pivotal role in initiating viral infection. Viral S1 and S2 subunit proteins are responsible for mediating attachment with ACE2 host cell receptors and membrane fusion, respectively. Herein, our main concern was to briefly shed light on a potential strategy involving disruption of SARS-CoV-2 S protein interaction with host cell receptors through design of neutralizing antibodies targeting RBD in S1 subunit, small peptide inhibitors, peptide fusion inhibitors against S2, host cell ACE2, and protease inhibitors, aiming to pave the way for controlling viral cell entrance.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.