
Abstract
Coronaviruses (CoVs) contribute significantly to the burden of respiratory diseases, frequently as upper respiratory tract infections. Recent emergence of novel coronaviruses in the last few decades has highlighted the potential transmission, disease, and mortality related to these viruses. In this literature review, we shall explore the disease-causing mechanism of the virus through human monocytes and macrophages. Common strains will be discussed; however, this review will center around coronaviruses responsible for epidemics, namely severe acute respiratory syndrome coronavirus (SARS-CoV)-1 and -2 and the Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Macrophages are key players in the immune system and have been found to play a role in the pathogenesis of lethal coronaviruses. In physiology, they are white blood cells that engulf and digest cellular debris, foreign substances, and microbes. They play a critical role in innate immunity and help initiate adaptive immunity. Human coronaviruses utilize various mechanisms to undermine the innate immune response through its interaction with macrophages and monocytes. It is capable of entering immune cells through DPP4 (dipeptidyl-peptidase 4) receptors and antibody-dependent enhancement, delaying initial interferon response which supports robust viral replication. Pathogenesis includes triggering the production of overwhelming pro-inflammatory cytokines that attract other immune cells to the site of infection, which propagate prolonged pro-inflammatory response. The virus has also been found to suppress the release of anti-inflammatory mediators such as IL-10, leading to an aberrant inflammatory response. Elevated serum cytokines are also believed to contribute to pathological features seen in severe disease such as coagulopathy, acute lung injury, and multiorgan failure.
Introduction
Coronaviruses (CoVs) are a group of enveloped viruses with single-stranded and positive-sensed RNA genomes. All seven presently identified that human coronaviruses have originated from animals such as bats and cause respiratory diseases (102). Most cause mild illnesses, such as the common cold, accounting for 15–30% of respiratory tract infections annually. However, several are capable of causing more serious illness such as severe acute respiratory syndrome coronavirus (SARS-CoV)-1, Middle East respiratory syndrome coronavirus (MERS-CoV), and the ongoing SARS-CoV-2 pandemic. These are zoonotic and highly pathogenic coronaviruses that have led to regional and global outbreaks.
Human coronavirus infections are generally associated with upper respiratory tract infections, of which the signs and symptoms commonly include fever, malaise, and cough; some patients may also develop lower respiratory tract infections. In contrast, SARS-CoV-1, MERS-CoV, and SARS-CoV-2 infections may remain asymptomatic in the early stage until severe pneumonia, respiratory failure, and/or sepsis develops, and even death. The fatality rate for SARS-CoV-1 is believed to be around 9.5%, 34.4% for MERS-CoV, and 2.5% for SARS-CoV-2. Those with certain risk factors, such as elderly over 50 years of age or immunocompromised patients, are associated with a much higher incidence of fatality (76).
Macrophages are key cells for host defense against pathogens and are abundant in all tissues of the human body. It is therefore relevant to review the current literature regarding the pathogenesis of human coronaviruses with a focus on the disease-causing mechanism of the virus through human monocytes and macrophages. These viruses have been found to alter innate immune response, which has significant implications for the prognosis and possible therapeutic interventions for the disease. This review shall look more in depth into human coronaviruses that are associated with significant mortality, namely SARS-CoV-1, MERS-CoV, and SARS-CoV-2, in consideration of the clinical impact of these strains.
Monocytes and Macrophages
Monocytes are key cells of the innate immune system and members of the mononuclear phagocyte system that comprises monocytes and two other major subtypes: namely, dendritic cells and macrophages. Monocytes are derived from hematological precursors in the bone marrow and enter the blood circulation, from which they are recruited into tissues throughout the body. These monocytes are capable of maturing into monocyte-derived macrophages within tissues (49). Current understanding of tissue macrophages indicates that many tissue-resident macrophages are self-renewing populations that arise from fetal progenitors and require minimal input from circuiting adult monocytes in a healthy environment (55,94).
The major functions of macrophages include antigen presentation, phagocytosis, and the release of cytokines (pro-inflammatory and/or anti-inflammatory mediators). Depending on the microenvironment, several factors can promote monocyte polarization. Specific stimuli include cytokines, growth factors, prostaglandins, and pathogen-derived molecules (5). Monocytic phenotypes represent a wide spectrum of activation states, which include the classical M1 (classically interferon [IFN]-γ-activated) and M2 (alternatively IL-4-activated) polarization states (56,84). M1-polarized macrophages are characterized by a high level of phagocytic activity and secretion of pro-inflammatory cytokines and chemokines, which induce Th1 activation and facilitate complement-mediated phagocytosis and type I inflammation. M1-polarized macrophages also carry out phagocytosis of microorganisms and matrix debris in the early stages of healing and have high antigen presentation capacity. M2 macrophages comprise a wide range of macrophage subtypes, such as tumor-associated macrophages, healing macrophages, and macrophages found in chronic inflammatory conditions; therefore, they play a crucial role in carcinogenesis and inflammation-dependent diseases (e.g., neurodegenerative disorders) (5,6). M2-polarized macrophages modulate Th2 response by producing anti-inflammatory mediators, leading to neutrophil, monocyte, and T lymphocyte recruitment. M2-polarized macrophages are highly endocytic, and they are involved in a variety of functions, including repair mechanisms, homeostasis, metabolic processes, and pathogenesis.
Macrophages possess high plasticity. Data from several studies have demonstrated that macrophages shift their polarization upon changes in the environmental conditions from M1 to M2 and vice versa (6,66). In the context of respiratory diseases, alveolar macrophages (AMs) are the key guardians of pulmonary homeostasis, playing important roles in modulation of inflammation and tissue repair (46). The recognition of damage- and pathogen-associated molecular patterns by pattern recognition receptors, such as C-type lectin receptors or toll-like receptors, allows them to recognize the presence of pathogens or products of injury and release pro-inflammatory cytokines (IFN-γ and TNF-α), which shift macrophage phenotypes toward M1 polarization (70). The protection offered by pathogen recognition is complemented by enhancing the presentation of antigens to T cells (4). In response to macrophage depletion, peripheral blood monocytes are recruited to the lung, where the microenvironment stimulates their differentiation into M1-polarized AMs (57,71). Once inflammation is controlled, tissue repair must take place to restore the normal tissue architecture. M2-polarized AMs are essential to orchestrate the repair of tissues through the release of anti-inflammatory cytokines, including IL-10 and TGF-β, and promote regulatory T cell response (4,38).
Currently, there is evolving evidence suggesting that resident AMs and recruited macrophages from the blood are key factors in the pathogenesis of acute lung injury (ALI) (26,51,68). ALI can be classified by significant hypoxemia, diffuse bilateral pulmonary infiltration, a decrease in pulmonary compliance, and a decrease in the functional residual capacity. Pathological changes include increased vascular permeability caused by alveolar-capillary membrane dysfunction, alveolar hemorrhage, and fibrin deposition (99). The pathophysiology involves an excessive inflammatory response to lung injury; accumulating neutrophils lead to overwhelming release of cytotoxic and pro-inflammatory mediators, and a depletion of AMs contributes to reduced regulation of inflammation (44). Alveolar hypoxia, a feature of ALI, has been associated with microvascular complications such as increased vascular permeability, generation of reactive oxygen species, and leukocyte–endothelial interactions. The causative mechanism is due to release of monocyte chemoattractant protein 1 (MCP1), or chemokine ligand 2 (CCL2), into circulation by hypoxic AMs. MCP1/CCL2 in turn triggers degranulation of perivascular mast cells, initiating the microvascular inflammatory cascade, as well as further recruitment of monocytes (12,13,15,34).
Human Coronaviruses
The CoV subfamily is genotypically and serologically categorized into four genera; the α, β, γ, and δ coronaviruses. The β-coronavirus can be further classified into four viral lineages, namely lineage A, B, C, and D. There are nearly 30 recognized CoVs that infect humans, mammals, fowl, and other animals. Human CoV infections (hCoVs) are caused by α- and β-CoVs (36). The common pathogenic coronaviruses are summarized in Table 1.
Comparison of Incubation Period, Commonly Affected Population, Fatality Rate, Target Cells and Receptors, Symptoms, and Pathological Hematological Findings Between Human Coronaviruses
ACE2, angiotensin-converting enzyme 2; 2APN, aminopeptidase N; DPP4, dipeptidyl-peptidase 4; MERS-CoV, Middle East respiratory syndrome coronavirus; SARS-CoV, severe acute respiratory syndrome coronavirus.
Coronaviruses were given their name based on the spike proteins on their membranes, which gives it a characteristic crown-like appearance. The virus structure comprises a nucleocapsid composed of genomic RNA and phosphorylated nucleocapsid (N) protein, which is enveloped by phospholipid bilayers and interspersed with two different types of spike proteins: the spike glycoprotein trimmer (S) that can be found in all CoVs and the hemagglutinin-esterase that only exists in a subset of β-coronaviruses. The membrane (M) protein and the envelope (E) protein are located among the S proteins in the virus envelope (28). The S protein plays a key role in viral adhesion to host cell receptors, and E protein has been found to promote pathogenesis in SARS-CoV-1 (74).
Pathogenesis of Human Coronaviruses
Coronavirus entry into monocytes and macrophages through antibody-dependent enhancement
In human coronaviruses, the initial attachment of the virion to the host cell is mediated by interactions between the S protein and its receptor. hCoVs then enter host cells through endocytosis or membrane fusion (60). The S protein–receptor interaction is the primary determinant for a coronavirus to infect a host species and also governs the tissue tropism of the virus. Many α-coronaviruses utilize aminopeptidase N as their receptor, SARS-CoV-1 and SARS-CoV-2 use angiotensin-converting enzyme 2 (ACE2) as their receptor, and MERS-CoV binds to dipeptidyl-peptidase 4 (DPP4) to gain entry into human cells, as summarized in Table 1.
The method of entry into immune cells, however, is likely to be antibody dependent. Peripheral monocytes and tissue macrophages lack ACE2 receptors required for SARS virus entry (89), while DPP4 receptors can be found on these cells (2,79). The entry mechanism of SARS-CoV-1 into macrophages is through antibody-dependent enhancement; in particular the anti-Spike protein antibodies and its interaction with the FcγRII (CD32) receptors and complement receptors on immune cells (50,96,98,104). The anti-spike protein antibodies form immune complexes with virions and mediate their attachment to Fc or complement receptors, allowing virus entry into monocytes or macrophages.
Currently literature has supported that SARS-CoV-1 could infect but could not proliferate in monocytes and macrophages, while MERS-CoV showed a greater ability to infect and replicate in these cells (50,60,92). Viral particles and genomic sequences were identified in both monocytes and lymphocytes in blood samples of SARS patients, implying that SARS-CoV-1 is capable of infecting both cell types. Approximately 50% of lymphocytes, with a majority being T cells, and 30% of monocytes in the circulation were infected by SARS-CoV-1 (37). HCoV 229E was likewise found to infect monocytes efficiently, induce apoptosis, and release high titers of infectious virus, whereas OC43 infected monocytes poorly and failed to induce increased apoptosis (103). The ability to infect monocytes and macrophages could provide hCoVs the mechanism to avoid host innate immunity and disseminate systematically (63).
Anti-spike protein antibodies skew macrophage polarization
A recent study performed on Chinese rhesus macaques revealed that in the context of SARS-CoV-1, the presence of S-IgG before viral clearance suppressed wound-healing responses and promoted MCP1 and IL-8 production and pro-inflammatory monocyte/macrophage recruitment and accumulation (62). The study also revealed that deceased SARS patients displayed similarly accumulated pro-inflammatory cells in the lungs and lack of wound-healing macrophages. Their sera also enhanced SARS-CoV–induced MCP1 and IL-8 production by human M2-polarized macrophages (62). The effect on M1-polarized macrophages was little to none. Other studies also suggest that anti-S specific antibodies have pathogenic effects in various animal models—multiple CoV vaccines were associated with an increase in eosinophilic pro-inflammatory pulmonary responses upon challenge of the immunized animals (8,40,47).
Another study found a temporal relationship between anti-spike protein antibody levels and the disease outcome in SARS-CoV-1 patients. While antibody activities in the deceased SARS patients were higher and developed faster compared to that in the recovered patients during the first 15 days after onset of symptoms, it diminished rapidly in the ensuing period of infection. Severe inflammation and significant higher levels of pro-inflammation cytokine were found in these deceased SARS patients (106). This is in agreement with the earlier finding that S-IgG response before viral clearance diminished anti-inflammatory response.
Spike protein induces phenotypic conversion of B cells into macrophage-like cells
Conversion of B cells into inflammatory-type macrophages can be readily induced to convert into functional macrophages with high efficiency, by the ectopic expression of transcription factor CCAAT-enhancer-binding proteins (C/EBP), and can be enhanced further by endogenous C/EBPα and β activation (9,20,25,100).
A murine study concluded that conversion of B cells to macrophage-like cells, similar to a pathophysiological response, could be mediated by a viral ligand, in particular the spike protein of SARS-CoV-1 (17). Severe local hypoxia, which is a condition often observed in afflicted lungs of SARS patients, also promotes this phenomenon (17). The study found that a proportion of transduced B cells expressed the activated monocyte/macrophage-specific markers Mac-1, CD19, and CD68 and became morphologically similar to monocyte/macrophages with enlarged and horseshoe-shape nuclei (17). This could potentially be a contributing factor to the massive accumulation of macrophages found in the lungs of CoV patients, which tend to exceed the amount of monocytes that can be provided by a patient's bone marrow within such a short time of virus infection (17).
Delayed interferon response
Macrophages are potent producers of type I IFNs and other pro-inflammatory cytokines that induce antiviral protection. IFNs mediate direct antiviral effects that limit viral replication by upregulating several well defined antiviral effectors, including PKR and RNaseL, while also modulating other aspects of the innate and adaptive immune responses through the induction of a wide array of IFN inducible genes (31).
SARS-CoV-1 and MERS-CoV infected macrophages show delayed but elevated levels of IFN and other pro-inflammatory cytokines (11,54,91). The delayed but excessive production of these cytokines and chemokines is thought to induce a dysregulated innate immune response to CoV infections. The findings have been consistent in murine models as well (10). Several CoV proteins are involved in antagonism of IFN response; nonstructural proteins (NSP) antagonize IFN pathway, such as NSP1 and papain-like protease (43,91). SARS-CoV-1 NSP7 and NSP15 have both been identified as potential IFN antagonists (32). Structural proteins such as the N protein of SARS and MERS CoV are capable of blocking type I IFN (64), and membrane (M) proteins block the transcription of IFN-β (11,22,86). However, hCoV HKU1 possesses M proteins as well, but they do not have the ability to suppress IFN response, suggesting that this ability is not conserved among all CoV strains (85). The studies are consistent with the finding that there was little to no induction of IFN-β in SARS-CoV-infected macrophages (16).
Another pathogenic feature of hCoV includes the inhibition of dsRNA sensors. Macrophages carry multiple dsRNA sensors designed to detect viral infection and amplify innate antiviral response. Coronaviruses, being positive-sense RNA viruses, will generate dsRNA intermediates during replication. Activation of these dsRNA sensors results in early induction of IFN, rapid apoptosis of macrophages, and therefore a protective immune response. However, some hCoVs can infect and propagate in macrophages without activating dsRNA sensors, such as MERS CoV (83). NSP4a, NSP4b, and NSP15 are some encoded proteins that have been found to inhibit dsRNA sensors (23,54,83). hCoVs reach high titers very early after infection (19,75,90) and have been found to produce multiple proteins that delay the IFN response, suggesting that an early antagonism of the IFN response might help hCoV delay or evade the innate immune response—this initially high viral load is also associated with a more severe prognosis in SARS (19).
SARS-CoV-2 has been found to activate limited to no type I IFN response from infected monocyte-derived dendritic cells (moDCs) and macrophages (MDMs), but triggered significant pro-inflammatory cytokine expressions in MDMs (7,101). Antiviral IFN and pro-inflammatory response were suppressed in moDCs through inhibiting STAT1 phosphorylation. The likely mechanism is through competitive binding of viral N protein to STAT1 with downstream kinases, thereby inhibiting their phosphorylation, a phenomenon exhibiting in SARS-CoV-1 as well (7,42,72); however, SARS-CoV-2 has been found to exhibit stronger inhibition (101).
Cytokine storm
The pathological finding of elevated cytokines and chemokines in hCoV patients is well documented. High serum levels of pro-inflammatory cytokines (IFN-γ, IL-1, IL-6, IL-12, MCP1, and TGF-β) and chemokines (CCL2, CXCL10, CXCL9, and IL-8) were found in SARS-CoV-1 patients with severe disease compared to individuals with uncomplicated SARS (11,91); the same can be seen in MERS-CoV and SARS-CoV-2 (65,82). Conversely, patients with severe disease had very low levels of the anti-inflammatory cytokines. IL-10, MCP1, and CXCL10 are notable for their strong chemoattraction of monocytes/macrophages, T cells, and acute inflammatory cells to the site of insult. Histopathological examination of lung tissues in SARS and MERS patients supports this, showing a high number of neutrophils and macrophages (14,88). The novel SARS-CoV-2 has also been associated with cytokine storm (97). Recent SARS-CoV-2 studies have revealed pyroptotic events in infected monocytes using NLRP3 inflammasome activation, contributing to release of pro-inflammatory cytokines through lytic monocyte death (29,80). Significantly elevated levels of monocyte-associated chemokines CCL2 and CCL8 have been observed, as well as CXCL2 and CXCL8, which function as chemotactic factors for neutrophils (7), consistent with clinical data showing elevated neutrophil levels in severe SARS-CoV-2 patients (58).
A cytokine storm can initiate a complex series of events; starting with the damage to epithelial and endothelial cells, promoting vascular leakage and thrombus formation that can progress to disseminated intravascular coagulation (DIC), and prolonged coagulopathy (27,35). Macrophage Activation Syndrome can also occur, leading to erythrophagocytosis and anemia (35). The combination of aforementioned pathologies may result in multiorgan failure. Figure 1 has summarized key aspects of the lethal pathogenesis of SARS-CoV-2.
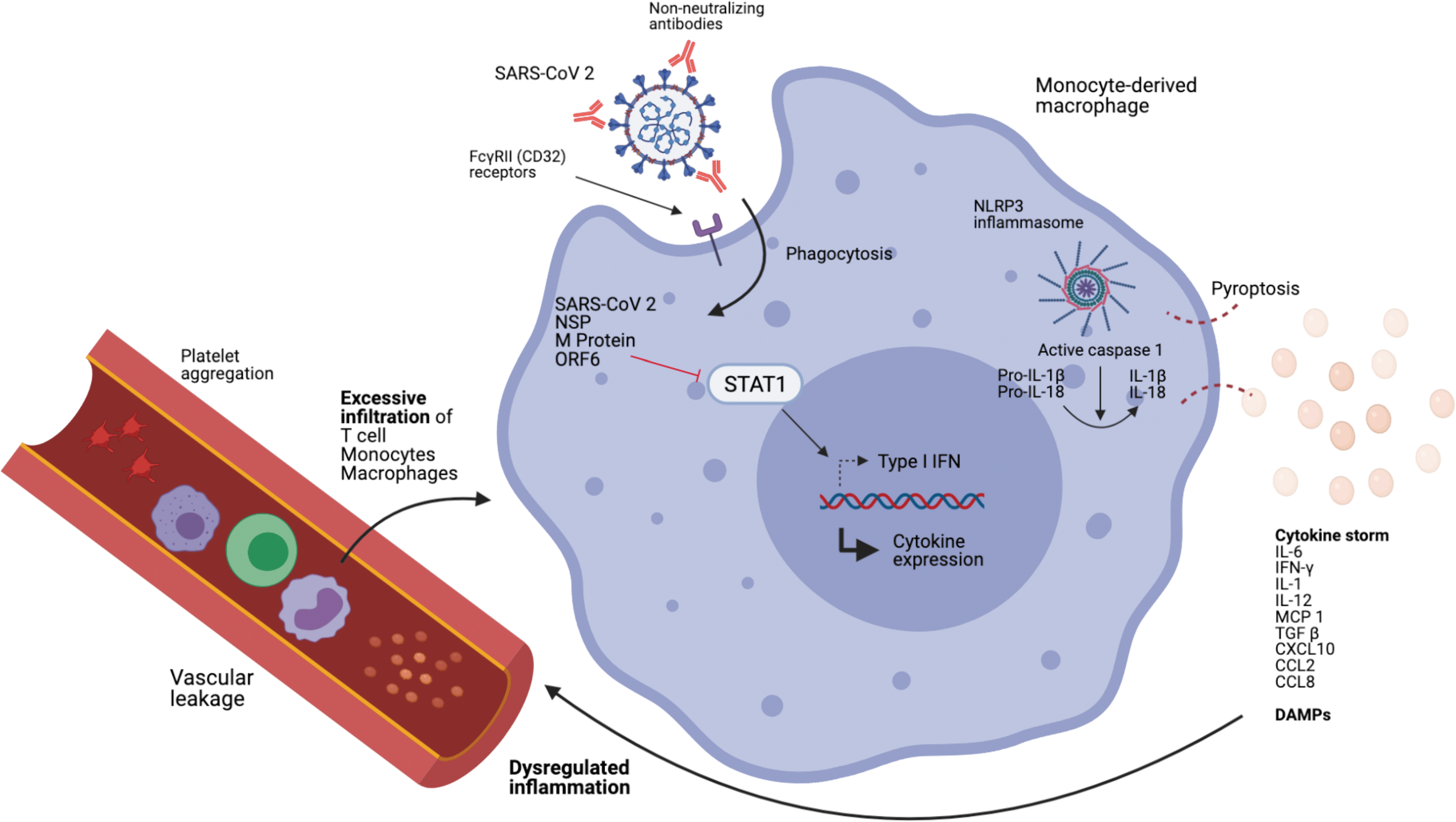
Diagram illustrating how SARS-CoV-2 can infect monocytes/macrophages and lead to an aberrant inflammatory response. The virus enters monocytes/macrophages through ADE. Viral NSP and M proteins are capable of inhibiting STAT1 nuclear translocation, which delays type I IFN response, leading to robust viral replication, while other cytokine expressions (namely CXCL10, CXCL5, TNF-α, IL-6, and CCL2) are upregulated. NLRP3 inflammasomes are activated in infected monocytes/macrophages leading to lytic cell death and the release of excessive pro-inflammatory cytokines and DAMPs. These chemotactic agents attract inflammatory cells and platelets, leading to excessive aggregation of inflammatory cells and coagulopathies seen in SARS patients. ADE, antibody-dependent enhancement; CCL2, chemokine ligand 2; DAMP, damage-associated molecular pattern; IFN, interferon; NSP, nonstructural proteins; SARS-CoV, severe acute respiratory syndrome coronavirus.
Discussion
Human coronaviruses utilize numerous mechanisms to undermine the innate immune response through its interaction with macrophages and monocytes. It is capable of entering immune cells, delaying IFN response, and thereby supporting robust viral replication. The virus also triggers the production of overwhelming pro-inflammatory cytokines, some of which attract other immune cells such as neutrophils and monocytes that can create a positive feedback loop, leading to aberrant inflammatory response. The elevated serum cytokines also contribute to pathological features seen in severe disease such as DIC, ALI, and multiorgan failure.
The virus has garnered much attention in the scientific community with several reviews looking at the interactions of monocytes and macrophages with SARS-CoV-2 (48,69). A review has further explored how the innate immune response, in unity, responds to the virus (93). Understanding the pathogenesis of the novel SARS-CoV-2, as well as previous strains, provides insight into possible therapeutic interventions for the current pandemic. Some experimental drugs, including chloroquine and hydroxychloroquine, are being trailed in SARS-CoV-2 patients, as it has been found to increase the pH of endosomes, which may limit the viral infection by inhibiting Cathepsin L, an enzyme that is vital for cleavage of the viral Spike protein, necessary for viral entry (24). In MERS CoV patients, chloroquine has been shown to inhibit replication in vitro (21). In addition to chloroquine's effect on CoVs, there is evidence of synergistic activity of chloroquine with glucocorticoids signaling by stabilizing glucocorticoid receptors (39). Since glucocorticoids are one of the recommended therapies for severe SARS patients, there is potential for chloroquine to be used in treatment of COVID-19 in combination with glucocorticoids, and trials are necessary to determine the efficacy of such combined therapy.
Glucocorticoid therapies were largely used during SARS-CoV-1 and MERS-CoV epidemic and are currently a popular immunomodulatory therapy option for SARS-CoV-2. In particular, dexamethasone is examined for its anti-inflammatory effects in SARS-CoV-2. However, corticosteroids are known to be immunosuppressive and, therefore, have been found to increase viral load in SARS-CoV-1 patients when administered early, leading to an exacerbated disease (11). Osteonecrosis was also an observed side effect in SARS-CoV-1 patients treated with high dose steroid pulse therapy (107). This implies that the timing, dosage, and duration of drug therapy are crucial in managing the patient and the disease. Recent evidence around the use of steroids has showed much promise (105) and will likely continue to be an integral management option of SARS-CoV-2 patients.
Cytokine storm phenomenon has been an area of keen interest in SARS patients. Novel research investigating inflammasome activity in SARS-CoV-2 patients has suggested NLRP3 inflammasome engagement and pyroptosis in human monocytes—exacerbating pro-inflammatory cytokine release (29,80). A study has further compared the efficacy of lopinavir, ribavirin, and atazanavir, with atazanavir showing potential in inhibiting early proteolytic processing of viral antigen and, thereby, preventing lytic monocyte death (29). This could suggest the value of investigating atazanavir boosted with low-dose ritonavir, which inhibits cytochrome P450 3A4 isoenzymes and prevents metabolism of protease inhibitors (52). Initial research using atazanavir and in vitro Vero cells and human pulmonary epithelial cells has shown promise. Atazanavir was found to impair SARS-CoV-2 replication and viral induced enhancement of IL-6 and TNF-α in infected monocytes (30), suggesting another potential drug for clinical trials. A key limitation, however, being that lopinavir had been similarly proposed for trials due to in vitro efficacy and preclinical studies, but clinical trials have shown no clinical improvement in patients (41). Nonetheless, atazanavir has shown to exhibit increased bioavailability in lung tissues and a high tissue affinity (33).
Tocilizumab, an IL-6 receptor antagonist, is on trial in various countries and a potential therapeutic option to manage excessively elevated pro-inflammatory IL-6, which has been linked to immunopathology and disease severity (53,61), and modulate cytokine storm syndrome seen in patients. Given that IL-6 plays a critical role in integrated innate and adaptive immune response against pathogens (18), inhibition could instead induce a slower viral clearance as supported by a recent prospective SARS-CoV-2 cohort study (67). It remains to be addressed whether IL-6's role in promoting chronic fibrotic disease and its inhibition in an acute disease phase could reduce long-term respiratory damage seen in SARS-CoV-2 patients (78). Therapeutic options for long-term tissue recovery due to severe SARS-CoV-2 remain to be explored, with biomaterials recently showing much potential in achieving regeneration of damaged tissues (81).
Conclusion
Coronaviruses have historically been responsible for numerous epidemics and the current pandemic. This review has consolidated existing literature surrounding existing coronaviruses' pathogenesis, summarizing the virus' abilities to trigger aberrant immune response through monocytes and macrophages. The virus is capable of infecting immune cells facilitating its dissemination, delay IFN response hence allowing robust viral replication, and triggering a cytokine storm leading to severe clinical manifestations. It is relevant in the current pandemic and provides insight into how patients' innate immune responses can be skewed leading to various clinical manifestations, as well as provides an opportunity to consider novel therapeutic options. The past is a good predictor of the future; the value of looking at various coronavirus strains also provides invaluable insight into similarities in the pathogenesis of this virus family, for better preparation of future novel strains—a grim question of when, not if.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Footnotes
Authors' Contributions
Chenghao was involved in research, reading, and selection of scientific articles to be included and the writer of the article.
Acknowledgments
Prof. Endre Kiss-Toth and Heather L. Wilson (University of Sheffield) were involved in forming the framework of the review and providing guidance on research and scientific writing.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.