
Abstract
Dengue is one of the most important vector-borne viral illnesses found in tropical and subtropical regions. Colombia has one of the highest rates of dengue cases in the Americas. Severe dengue virus (DENV) infection presents with capillary leakage, hemorrhage, and organ compromise, eventually leading to death. Over the years, there have been many efforts to develop a vaccine that guarantees protective immunity, but they have been partially successful, as such immunity would need to guarantee protection against four distinct viral serotypes. Absolute platelet count is a laboratory parameter used to monitor the clinical progression of DENV, as infection is often accompanied by thrombocytopenia. Although this finding is well described with respect to the natural history of the disease, there are various hypotheses as to the cause of this rapid decrease, and several in vivo and ex vivo models have been used to explain the effect of DENV infection on platelets and their precursors. DENV infects and activates platelets, facilitating their elimination through recognition by phagocytic cells and peripheral margination. However, infection also affects the precursors in the bone marrow by modulating megakaryopoiesis. The objective of this article is to explore various proposed mechanisms of DENV-induced thrombocytopenia to better understand the pathophysiology and clinical presentations of this highly relevant viral infection.
Introduction
Dengue virus (DENV) infection is associated with high morbidity and mortality among both children and adults in tropical and subtropical regions. DENV is a public health issue, as it is transmitted by Aedes mosquitoes (primarily Aedes aegypti) and therefore can impact a high percentage of the population, imposing a large burden on health care systems (39). During 2019, the incidence of dengue in the Americas was >321 dengue cases per 100,000 people. A total of 1,538 deaths were also reported and the highest recorded number of infections in the history of dengue in the region (119).
Early clinical management is fundamental for avoiding progression to the severe form of the disease. However, there is currently no specific antiviral treatment available. Vaccination has been proposed as an effective preventative strategy. In 1944 and 1945, Kimura and Hotta (45) and Sabin and Schlesinger (97) developed the first monovalent vaccines. At present, the only DENV vaccine approved in some countries for use in humans is Dengvaxia®, a chimeric tetravalent vaccine with a live virus (CYD-TDV) that uses yellow fever viral strain 17D as a base, with recombinant genes—premembrane (PrM) and envelope (E)—along with each of the four DENV serotypes (42). This vaccine has been controversial owing to differences in its efficacy against different viral serotypes and an increase in the risk of severe illness when given to young children previously unexposed to DENV (43,109).
There are other vaccines in the final phases of clinical trials. One of these is the Takeda vaccine candidate (TAK-003), a vaccine that consists of a DENV-2 virus base with chimeric DENV-DENV (53,117). Other vaccines include a live attenuated (LAV Delta 30), DENV180 protein subunit (DEN1-80E), and DENV DNA vaccine, also in advanced stages of study (32,33).
In recent decades, there have been continued efforts to understand and treat DENV infection. Although it is accepted that prevention and management of the infection are key, it is also fundamental to understand the impact of DENV at the cellular and tissue levels. Accordingly, observational and experimental studies have explored this topic (3,34,48,70,84). The current classification of DENV infection uses not only clinical signs and symptoms but also certain laboratory parameters such as hepatic enzymes. Laboratory tests may be used for diagnostics or to follow the disease course (116). For example, a decrease in absolute circulating platelet count has been correlated with severe forms of the disease in animal models and humans (71,82).
A fast and significant decrease in the absolute count number of circulating platelets is a common pathophysiological phenomenon with clinical consequences during DENV infection (56,116). Thus, it has been proposed that this specific cell type is a preferential direct and indirect target of viral infection (80,100). Several mechanisms, including the effects of DENV infection on bone marrow platelet precursors or peripheral vascular consumption of mature platelets, have been proposed to explain the decrease in the absolute number of this cell type in circulation during DENV infection (6,88). The objective of this article is to discuss the current state of knowledge about DENV-platelet interactions and the possible causes of thrombocytopenia during DENV infection.
Can DENV Infect Human Platelets, and How Does This Affect Its Pathogenesis?
The first attempts at the viral infection of platelets were conducted in the 1950s and consisted of exposing human platelets to influenza virus inducing their agglutination (26,73) and then, enabling the capture of the first images of viral infection of platelets using electronic microscopy (30).
Human platelets are small cytoplasmic fragments ranging 2–4 μm in diameter derived from bone marrow megakaryocytes. They travel through blood vessels and contribute to primary hemostasis and subsequent coagulation through activation and aggregation. Mitochondria, endoplasmic reticulum, lysosomes, and between 50 and 80 granules are found in the cytoplasm of each platelet (36). The granules can be grouped into three major types: α-granules, dense granules, also called δ-granules, and lysosome-like granules. The α-granules contain proteins such as chemokines, cytokines, and growth factors necessary for platelet function. They contain small molecules such as adenosine diphosphate (ADP), serotonin, polyphosphates, glutamate, histamine, and calcium, which are necessary for hemostasis. The third type of granule functions similar to lysosomes, which vary in molecular cargo, biogenesis, trafficking, and exocytosis. Other organelles present in platelets include peroxisomes and T granules, characterized by the upregulation of Toll-like receptor 9 (TLR9) during pro-platelet production (36,110). Furthermore, platelet lysosomes contain glycohydrolases along with enzymes that degrade glycoproteins. Although the principal role of platelets is to maintain hemostasis, they also play an important role in other pathophysiological processes, such as tumor growth, metastasis, inflammatory response, and host defenses, as they are an important source of cytokines (19,60).
The presence of DENV in platelets and its capacity to enter these cells have been studied for years. It has been confirmed that platelets can bind and internalize infectious agents (19,20,41). However, the impact of DENV on these cells is only partially known. The recognition of viral pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs) facilitates the production of interferon (IFN)-I, whose stimulating antiviral and immune properties are beneficial for viral elimination. In the short term, IFNs impact hematopoiesis, resulting in temporary aplasia and potential alteration of megakaryopoiesis. This decrease in the regulation of hematopoiesis is likely a protective mechanism of the microenvironment that limits damage to the marrow compartment or progenitor cells during the process of elimination of infected cells (74,83).
In vitro models have been used to identify platelet mechanisms of viral binding and internalization through proteoglycans such as heparan sulfate and DC-SIGN, a C-type lectin receptor. It has been shown in human platelet precursors in vitro that once the virus has entered the cells, it begins with a replicative cycle and the translation of viral proteins, thus continuing to replicate its genome, and finally assembling new virions (8).
Once infected, platelets increase the expression of cell membrane proteins such as CD40 ligand (CD40L), CD42b, CD62P, and major histocompatibility complex class I (MHC-I). Evaluation of these interactions using primary human platelets and DENV serotype 2 (DENV-2) has shown a significant increase in CD40L in the culture supernatant and greater secretion of cytokines such as granulocyte-macrophage colony-stimulating factor, interleukin (IL)-6, IL-8, IL-10, and tumor necrosis factor (TNF-α) compared with controls (77).
One method of confirming viral replication, apart from genome amplification (positive or negative single-strand RNA) using real-time RT-PCR (RT-qPCR), is through the detection of structural and nonstructural proteins. Among these, nonstructural protein 1 (NS1) has emerged as a useful alternative. This protein is not present in the infecting viral particle, but it is expressed and actively secreted by infected cells. Although it is known to be essential for viral replication, the exact mechanisms through which it participates in this process are not entirely known (24). NS1 is also a protein highly utilized in current diagnostic evaluation.
Furthermore, it is possible to isolate platelets in peripheral blood for subsequent in vitro infection with DENV. Methods such as RT-qPCR can then be used to quantify viral RNA and western blots can be used to identify the NS1 protein, confirming replication and translation of the viral genome in thrombocytes (100). However, it has also been observed that infectious viral particles do not accumulate in platelets or their precursors in culture (50). Previous findings suggested that although platelets support DENV replication, they do not assemble or release even very low titers of infecting viral particles (50,100).
It is not clear whether platelets contribute to the transport and dissemination of DENV infection in vivo or if, on the contrary, they aid in host defense against the infection. For now, the debate continues, and the hypothesis has been proposed that the DENV-platelet interaction may have a dual role depending on the infecting serotype and the environment in which platelets and megakaryocytes are found (35).
Platelet activation has also been reported in human natural DENV infection, as demonstrated by the increase in expression of membrane proteins such as platelet integrin receptor (αIIbβ3), lysosomal marker CD63, and granular marker CD62P (P-selectin) (71).
Platelets are involved in the pathophysiology of DENV infection through partially described mechanisms. First, as immune cells, platelets enable the host immune system to activate α-granules, which release cytokines such as platelet factor 4 (PF4), RANTES, platelet-derived growth factor, and transforming growth factor-beta (TGF-β) (65). Second, thrombocytes can impact the integrity of endothelial cells that coat blood vessel walls, because platelets and their precursors secrete vascular endothelial growth factor A (VEGF-A), which alters the phenotype of endothelial cells, thus increasing vascular permeability. This activation explains in part the capillary leakage classically observed in patients with severe forms of DENV infection. Significantly increased circulating levels of VEGF-A have been found in patients with more severe forms of the illness (38,64,107).
The rapid and transient thrombocytopenia and subsequent recovery observed in DENV infection suggest that short-lived immune factors such as cytokines are responsible, at least partially, for this process (102). DENV infection induces a significant systemic cytokine response secreted from innate and adaptive compartments, and the rapid and transient increase in cytokines has been proposed as a mechanism related to endothelium activation, vascular leakage, and severe clinical forms of DENV infection (14,18,103). Some of these cytokines, such as IL-8, IL-10, TNF-α, and soluble receptor-II (sTNFRII) (40,58), have been negatively correlated with the platelet count.
The application of recombinant human IL-10 to healthy adults resulted in thrombocytopenia. This effect was associated with a decrease in the number of megakaryocytes in bone marrow rather than a decrease in platelet half-life owing to peripheral sequestration (101). In addition, the application of TNF-α induces endothelial damage with platelet aggregation mediated by von Willebrand factor (vWF) and adhesion molecules in a human melanoma model (92). Caspase activation (Caspase 3, particularly) has been observed in murine platelets exposed in vivo or in vitro to TNF-α, a finding that supports cell death as a mechanism involved in TNF-α-mediated thrombocytopenia (85). Thus, TNF-α is one of the cytokines clearly associated with thrombocytopenia through a decrease in platelet half-life in the periphery.
Nevertheless, many questions remain unanswered regarding thrombocytopenia induced by DENV infection, such as the in vivo contribution of the spleen and endothelium or the mechanisms of thrombocytopenia during primary or secondary infection (57). Purified primary platelet models have been used for decades and have been useful for elucidating direct viral effects on these cells. However, they have important limitations, such as platelet activation induced by the purification process.
New models for the study of thrombocytopenia in DENV are needed (12). The development of both immunodeficient and immunocompetent murine models will be key. As has been evaluated in models of autoimmune thrombocytopenia, adoptive transfer from infected individuals could provide additional information on humoral and cellular factors responsible for the drop-in platelet numbers during DENV infection. In addition, the analysis of platelet functionality through evaluation of its aggregation capacity is another important source of information on the role of platelets in DENV infection (111).
Effects of DENV Infection on Platelet Precursors
Another effect of DENV infection is bone marrow suppression. The reduction of megakaryopoiesis and granulopoiesis in the first 4 days of infection accounts for the decrease in leukocyte and platelet counts in peripheral blood (13). Once the critical phase of the illness has passed, recovery of the absolute cell count occurs notably in conjunction with clinical improvement, as hyperplasia of megakaryocytes, myeloid cells, and erythrocytes has been observed after the 6th day following symptom onset (76,114). Understanding the causal mechanisms of the bone marrow changes and their impact on thrombopoiesis is key to understanding DENV pathology, offering an opportunity to impact both diagnosis and management and, therefore, the patient's outcome.
Hematopoietic stem cells and megakaryocytes represent ∼0.4% of the total cellular contents of bone marrow; as a result, these cells are difficult to isolate and study. The use of cell lines derived from these types of precursors, which partially resemble the conditions observed in vivo, has been considered an alternative to evaluating the interaction between DENV and human platelet precursors in vitro.
During the initial stages of platelet progenitor maturation, glycoproteins GPIIb (CD41a) and GPIIIa/β3 (CD61) are early markers of megakaryocytes (MK) differentiation (63). At this time, they are the most abundant proteins on the platelet precursor surface and they form a functional dimeric complex that recognizes and binds to fibrinogen and vWF (94). During the course of maturation, they start to express a second vWF receptor, which consists of three chains called GPIb α, GPV, and GPIX, known in conjunction as CD42b, a marker that is commonly used to differentiate between immature and mature MKs (63,99).
Megakaryocyte and erythrocyte precursors have been described taking a role in the immunity during DENV infection (17). There is evidence of changes in the morphology and proportions of the cell populations in bone marrow infected with DENV (75). The first reports by Bierman and Nelson showed that patients with severe dengue had few to no MKs during hospitalization per analysis of their bone marrow aspirate. Then, during the convalescent phase, both their immature and mature MK counts had recovered (13), which gave rise to further studies that have confirmed that DENV infection is accompanied by a decrease in central thrombopoiesis in human and animal models (76,87). These observations are most consistent with early suppression of megakaryocytopoiesis in the bone marrow (76).
The primary precursor cells necessary to explore these theories must be obtained using bone marrow aspiration—a painful and invasive procedure. Thus, many studies have used components of peripheral blood, which are more practical and easier to obtain. One study reported a positive correlation between viral load in the plasma of patients with acute dengue and the circulation of CD61-positive cells (an MK marker), as well as with an increase in IFN-γ levels in serum (46).
MK and megakaryocytic cell lines respond to viral infections and recognize viral PAMPs, thus secreting high levels of IFN-α and β (4), which reduce the production of platelets in vitro through autocrine IFNAR signaling. Apart from the reduction in platelet production, it is expected that the direct infection of MK, the involvement of PRR, and locally induced cytokine signaling change the phenotype of platelet progeny generated during the infection; this would influence immune and inflammatory processes mediated by platelets in the periphery (51).
In vivo studies, starting with modeling infection through viral inoculation of humanized mice and using quantification of viral RNA and flow cytometry to detect intracellular expression of E viral protein in bone marrow samples, have shown that 1.5% of immature human megakaryocytes and 35% of mature megakaryocytes become infected with DENV during infection (112). Furthermore, the pattern of selective expression of membrane proteins is necessary for DENV to infect this cellular population, thus affecting the dynamics of thrombopoiesis.
Models of MK and platelet infection have been proposed ex vivo. Hematopoietic precursor cells from the umbilical cord stimulated with a cocktail of cytokines consisting of thrombopoietin, IL-9, IL-6, and SCF, differentiate into immature MKs. After treatment with DENV, these cells maintained a constant level of viral RNA that persisted, but did not increase, over time (112).
In vitro studies have advanced our understanding of the impact of infection on host cells. The use of cell lines provides an opportunity to analyze the impact of viral infection: MEG-01 cells have been particularly useful, as they are megakaryoblastic cells obtained from the bone marrow of a patient with chronic myelogenous leukemia in blast crisis (78). DENV-2 propagates efficiently and produces viral particles when infecting this cell line (28). Infection has been detected through the presence of viral RNA inside MEG-01 cells, confirming that these cells are susceptible to DENV-2 virus infection (8). This cell line differentiates into mature platelets under certain conditions, making it an attractive in vitro model to study the effect of DENV infection on thrombopoiesis.
One characteristic of platelet progenitors that appears to be influenced by the infection is the endomitosis, where mitosis is interrupted in the middle of anaphase and the cell enters a new mitotic cycle. As a consequence, the number of chromosomes increases inside a single-lobed nucleus, resulting in polyploidy. This condition allows for the amplification of the cellular genome to support the synthesis of the proteins necessary for cellular growth and the formation of platelets. Thus, DENV infection disturbs platelet formation and may contribute to the genesis of thrombocytopenia (5). The production of platelet-like particles derived from MEG-01 cells has been demonstrated through treatment with phorbol 12-myristate 13-acetate (PMA), a potent activator of protein kinase C (PKC) (106).
Owing to the multiple actions of PKC isoforms, the mechanisms of PMA-induced differentiation of MEG-01 cells are very diverse. In general, it is used to recreate particles similar to mature platelets in vitro from the MEG-01 cell line. These particular cells show an increase in β1-tubulin, which is important for the late phase of MK differentiation and the early phase of circulating platelet production. Therefore, PMA-treated MEG-01 cells are considered a valid model to study the process of platelet production (47).
The maturation stage of platelet development in the context of DENV infection appears to influence the cellular capacity of viral replication. Existing theories propose that differentiation of platelet progenitors results in increased replication of DENV, as a greater number of viral RNA copies are observed in MEG-01 cells that have been infected and then differentiated with PMA than in those that do not undergo differentiation. Nonstimulated MEG-01 cells are susceptible to DENV infection, whereas MEG-01 cells that are differentiated and prestimulated with PMA are refractory to infection and viral replication (8). In conclusion, DENV infects primary and cell line-derivate platelet progenitors and affects the process of megakaryopoiesis, which directly, or in combination with other factors, results in the decrease in platelet counts as observed in patients with dengue.
What Explains Thrombocytopenia in DENV Infection?
An understanding of the complex interactions between the virus, platelets, and other cellular populations is necessary to understand the context of DENV infection. Platelets and leukocytes are fundamental for primary hemostasis and coagulation. The leukocyte–platelet interaction is modulated by various proteins, such as CD42 (glycoprotein GPIb) on platelets, macrophage-1 antigen (MAC1, also known as αMβ2), complement receptor 3 (CR3), CD11b, and integrin α-M (ITGM). Once interactions are established, phagocytic cells are recruited to the inflamed endothelium, where they identify damaged or altered thrombocytes and eliminate them (11).
It has been shown that isolated platelets from the peripheral blood of patients in the acute phase of DENV infection are activated and trapped by monocytes, demonstrating a role of monocytes and macrophages in DENV thrombocytopenia (34,98). However, when inactivated by prostaglandin E2 (PGE2), phagocytosis is halted (37), as PGE2 inhibits platelet activation and aggregation, thus inhibiting various stages in the activation of the arachidonic acid metabolic cascade. This cascade normally activates adenylyl cyclase, increasing the levels of AMPc, a crucial mediator of platelet aggregation (29). In turn, both this signaling pathway and the consequent cellular interactions are impacted by DENV infection of platelets.
The host immune response results in decreased peripheral platelet counts in DENV-infected patients through several partially understood pathways. Three complementary and not mutually excluding theories have been proposed (Fig. 1): A. A decrease in platelet production as a consequence of selective suppression in bone marrow (Fig. 1A). B. An increase in peripheral platelet consumption (Fig. 1B). C. Platelet lysis occurring through the formation of immune complexes (9) (Fig. 1C).
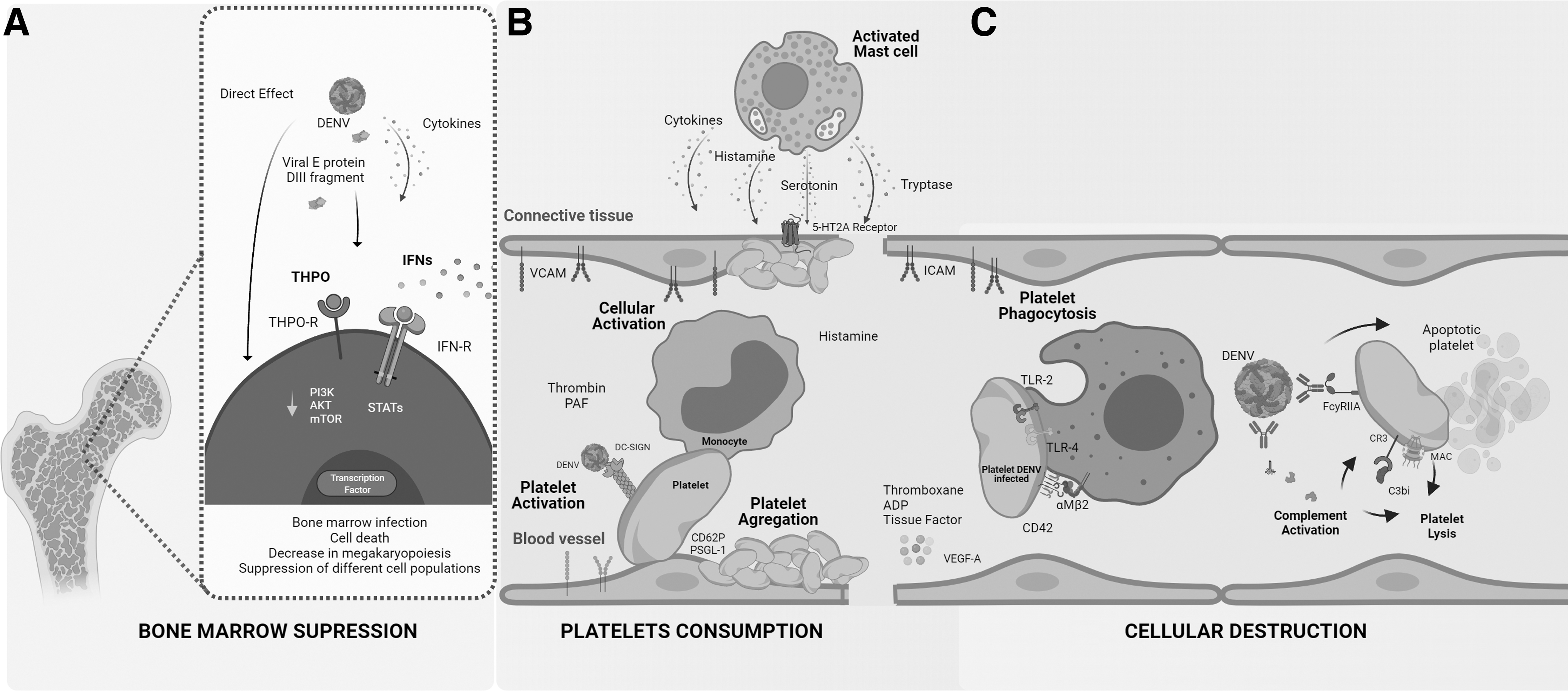
Cellular and molecular interactions contributing to thrombocytopenia during DENV infection.
DENV can directly infect and induce cell death of platelet precursors in bone marrow (10) (Fig. 1A). In addition, the application of domain III fragment from the E protein induces suppression of mice and human platelet precursors in vivo and in vitro, respectively (59), demonstrating that viral replication or whole dengue viral particles are not a necessary requirement for bone marrow suppression (Fig. 1A). Immune factors released in response to viral infection such as IFNs can induce significant effects the megakaryopoiesis.
Various studies in human and murine models have demonstrated that alterations in the signaling pathway of IFNs contribute to the depletion of hematopoietic progenitors, thus affecting the signaling pathways of canonical and noncanonical transcription factors, which may act as a myeloid differentiation checkpoint in hematopoietic stem and progenitor cells (68) (Fig. 1A). In support of this, rapid and remarkable thrombocytopenia depending on the type I IFN expressed in bone marrow is induced after injection of TLR agonists in vivo (93).
It has also been proposed that a decrease in IFN-γ levels results in depletion of stem cells and therefore suppresses marrow function (68,72). A simultaneous decrease in granulocytopoiesis and megakaryopoiesis is a strong indicator of marrow dysfunction at the level of hematopoietic pluripotent stem release cells or that the virus may simultaneously affect progenitors of two or more cell lineages. Recently, other new signaling pathways highly related to megakaryopoiesis such as the PI3K/AKT/mTOR axis have been shown to be modulated by DENV infection (55) (Fig. 1A).
In support of the theory of peripheral platelet consumption (Fig. 1B), patients with DENV infection may develop disseminated intravascular coagulation (DIC), a condition caused by the continual activation of the coagulation cascade and fibrinolytic system. In DIC, circulating platelets are activated by multiple factors, such as thrombin, which then release a variety of immune-stimulating molecules, such as histamine and platelet-activating factor, as well as coagulation cascade activators, such as thromboxane, ADP, and tissue factor (Fig. 1B). The consumption of procoagulant factors contributes to one of the frequent complications of infection—hemorrhagic syndrome—which is one definition of a severe presentation of the illness (31,116).
The other mechanism proposed to explain thrombocytopenia during DENV infection involves antibody-mediated peripheral destruction of platelets (Fig. 1C). A unique feature of DENV is that it has four different serotypes. Although these serotypes are structurally related, the generated protector host response is homotypic; that is, infection with one serotype does not generate long-lasting protection against the other three (44).
However, during secondary DENV infection, preexisting antibodies—particularly those against viral proteins prM and E—facilitate efficient binding of immature and mature viral particles to cells that express Fc receptors (95). Platelets express the FcγRIIA receptor (Fig. 1C). The binding of anti-prM-DENV complexes makes platelets susceptible to immune destruction. Platelet–immunoglobulin M or platelet–immunoglobulin G interactions are also important to explain DENV-mediated thrombocytopenia and the more severe presentations of this disease, such as those occurring during secondary DENV infection (79).
On the contrary, platelet-DENV complexes can bind to C3, IgM, and IgG, which subsequently facilitate their elimination through immune pathways (6,80,113) (Fig. 1C). It is important to take into account that the functions of C3 fragments and IgG include cellular opsonization and clearing (89). Flow cytometry has been used to evaluate proteins bound to the platelet membrane in supernatant from patients with acute dengue infection, showing an increase in binding of these proteins to the platelet membrane, which coincides with the timing at which the platelet count is the lowest, as recorded in the peripheral blood during days 4 and 6 of the disease course (80).
As the time course of the infection continues, platelets are activated and consequently decrease in count owing to recognition by phagocytic cells (Fig. 1C). Viral proteins on the membranes of infected platelets are recognized through interaction with Toll-like receptors 2 and 4 (TLR2, TLR4), which facilitate activation, aggregation, and adherence to endothelial cells and phagocytosis through macrophages (7,22) (Fig. 1C).
DENV can also activate platelets through lectin type C receptors, such as SLK 2, which at the same time stimulate macrophages and neutrophils through the CLEC 5A and TLR2 pathways (104,105). Neutrophils are then activated, forming neutrophil extracellular traps (NETs), which favor prothrombotic cells such as platelets, red blood cells, and molecules involved in intrinsic and extrinsic coagulation (89,104). NETs also activate platelets through TLR4, which generates a positive feedback circuit of additional NET and platelet activation.
Other proinflammatory and coagulation cascade-related molecules that interact with platelets are induced during dengue infection. After DENV infection, several cell lines and human primary cells show increased expression of the high mobility group box1 protein (HMGB1) (25,49,81), a nuclear damage-associated molecular pattern (DAMPs) released during necrosis and cell damage (118). Platelets are one of the main sources of HMGB1, and HMGB1 is also a strong inducer of platelet activation (96). Platelet activation through TLR by HMGB1 induces their aggregation by increasing peripheral sequestration. In addition, platelet-derived microvesicles containing HMGB1 have been shown to be a strong inducer of NETs (69), further increasing peripheral platelet loss.
A regulatory mechanism of the proinflammatory effects of HMGB1 is its uptake by thrombomodulin, an antithrombotic glycoprotein with anti-inflammatory effects that is expressed in endothelial cells and monocyte–macrophages (1). Although both circulating HMGB1 and thrombomodulin are increased in naturally infected patients with DENV and have been associated with the clinical severity of infection (2,16), their role in thrombocytopenia in dengue is not clear.
The role of mast cells and their secreted products has suggested a new and interesting mechanism of DENV-induced thrombocytopenia (Fig. 1B). After in vitro treatment with DENV immunocomplex, human cord blood-derived mast cells and mast cell lines release cytokines such as TNF-α that induce expression of adhesion molecules such as vascular cell adhesion molecule-1 on the surface of endothelial cells (15) (Fig. 1B). Using mice deficient in mast cells and mast cells deficient in tryptophan hydroxylase-1 (an enzyme necessary for the production of serotonin in mast cells), strong platelet and endothelial activation mast cell dependent, with aggregation and a decrease in platelet number observed (66).
This effect is mediated through the 5HT2A receptor (Fig. 1B), because drugs that selectively inhibit this receptor attenuated the thrombocytopenia (66,67). The increase of vascular permeability is a key clinical marker of severe dengue. Enzymes released after mast cells degranulation such as tryptase, have been associated with plasma leakage through disruption of endothelial tight junctions (91) (Fig. 1B). These findings indicate possibly new therapeutic targets.
Treatment of Thrombocytopenia During DENV Infection
At present, there is no specific antiviral treatment for infection with DENV, and treatment is focused on aleviating symptoms and treating clinical events during the infection, especially those that occur during the critical phase. Previous efforts have addressed the treatment of thrombocytopenia during DENV infection. It is now clear that there is no correlation between thrombocytopenia and the risk of bleeding during DENV infection (27,86), although there is a relationship of thrombocytopenia with the severity of the disease.
Therapeutic strategies have been developed for the prevention or treatment of bleeding related to thrombocytopenia during infection with DENV. To date, platelet transfusion has been the most studied. Prophylactic platelet transfusion in patients without bleeding has not shown significant reduction in the risk of developing subsequent bleeding. In addition, it is an expensive procedure with a high frequency of adverse events (23,62,115), such as urticaria, maculopapular rash, and pruritus (61). In addition, a high percentage of patients, usually those with lower platelet counts, do not respond or respond poorly to transfusion (52). Thus, there are clear indications for platelet transfusion in DENV infection based on a low platelet count with uncontrolled continuous bleeding and corrected coagulation factors, applying the same approach to patients with severe thrombocytopenia who must undergo a surgical procedure for other causes (108,116).
Other measures for the treatment of thrombocytopenia, such as intravenous immunoglobulin or thrombopoietin receptor agonists, have shown positive results, although strong evidence is still lacking in this regard (21,54,90). The above-mentioned limitations and lack of evidence support the need for controlled clinical trials for the treatment of bleeding associated with thrombocytopenia in patients with DENV infection.
The mechanisms summarized in this study contribute not only to understanding of thrombocytopenia but also to understanding the hemorrhage and capillary leakage, both critical clinical characteristics of DENV infection.
Conclusions
Platelets present in peripheral blood are a cellular subpopulation particularly impacted by DENV infection. DENV-platelet interactions are only partially understood, and many efforts over the last few years have made progress in understanding the pathophysiology and identifying the causal mechanisms of thrombocyte alterations. Three theories have been proposed to explain thrombocytopenia in DENV infection: selective bone marrow suppression, an increase in peripheral consumption, and immune complex-mediated elimination. Furthermore, DENV infection not only affects platelet counts but also activates these cells on a peripheral level and initiates several changes at a central level.
It is important to recognize that platelet infection impacts their functionality and can consequently induce alterations in hemostasis. It is also necessary to understand DENV infection as a multifactorial vascular illness, where a combination of immune system activation, coagulation, expression of anaphylatoxins and inflammatory proteins, and platelet sequestration and dysfunction results in a strong host response. Additional studies are necessary to explore the immunopathogenesis of DENV infection and to develop more options for diagnostics and treatment of a disease that, every year, takes the lives of thousands of people around the world, despite public health efforts to control the spread of the vector.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Paula Ximena Losada is a doctor and young researcher in the Division de Inmunología, Programa de Medicina, Facultad de Salud, Universidad Surcolombiana and was financed by the Ministerio de Ciencia, Tecnología e Innovación de Colombia, convocatoria 856-2019, “Jóvenes Investigadores e Innovadores para el Departamento del Huila” (Convenio No.80740-804-2019). Carlos F. Narváez was funded by the Sistema General de Regalías del Departamento del Huila—Minciencias (Grant 2020000100145).