
Abstract
Restoration of exhausted hepatitis B virus (HBV)-specific CD8+ T cells is one of the important strategies for inhibition of viral replication. The role of interleukin (IL)-33 to recovery of CD8+ T cell activity is not fully elucidated. We investigated the effect of IL-33 on viral-specific CD8+ T cell responses in chronic hepatitis B (CHB) patients in vitro by both phenotypic and functional analysis. Plasma IL-33 was downregulated in CHB patients, while effective antiviral therapy rescued IL-33 expression. There was no significant difference of IL-33 receptor mRNA relative level in CD8+ T cells between CHB patients and controls. IL-33 induced the proliferation of HBV-specific CD8+ T cells, and reduced programmed death-1 expression on HBV-specific CD8+ T cells. IL-33 promoted the direct cytolytic activity of CD8+ T cells against HepG2.2.15 cells through boosting perforin and granzyme B production. Furthermore, IL-33 administration increased HBV-specific CD8+ T cell-mediated HBV replication and HBV antigen secretion mainly via enhancement of interferon-γ and tumor necrosis factor-α. IL-33 reinvigorated antiviral activity of HBV-specific CD8+ T cells, revealing that IL-33 might contribute to viral clearance in persistent HBV infection.
Introduction
The outcome of hepatitis B virus (HBV) infection is closely associated with the interaction between viral replication and host immune response (20). Acute HBV infection always results in proliferation of polyclonal and multispecific cytotoxic T cells, leading to not only the viral clearance and acute self-limiting infection but also severe liver injury (9). In contrast, chronic HBV infection reveals dysfunctional or exhausted immune status. In chronic hepatitis B (CHB) patients, CD8+ T cells secreted insufficient antiviral cytokines and cytotoxic molecules, such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), perforin-1, and granzyme B, indicating a decreased cytotoxicity to HBV-infected hepatocytes (12).
Moreover, immune checkpoint molecules, including programmed death-1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), T cell immunoglobulin domain and mucin domain-3 (TIM-3), and lymphocyte activation gene-3 (LAG-3), were found to be increasingly expressed in CD8+ T cells in CHB patients, further suggesting the state of immune exhaustion during persistent HBV infection (4,10). Thus, recovery of exhausted HBV-specific CD8+ T cells is one of the important strategies for functional cure for hepatitis B (24).
Interleukin (IL)-33 is a member of IL-1 cytokine family, and signaling through IL-33 requires the binding to its receptor suppression of tumorigenicity 2 (ST2) (5). IL-33 is a nuclear cytokine, which is mainly expressed by endothelial and epithelial cells, plays a dual function in both physiological and pathological condition (19). IL-33 could also be secreted by activated immune cells, and is necessary for Th2-induced airway inflammation (25). IL-33 exacerbated influenza virus-induced asthma by enhancement of airway hyperresponsiveness and inflammation through halting innate and adaptive immune response as well as inhibiting cytolytic antiviral activities (29). In contrast, IL-33 could suppress HBV replication through ST2 receptor mainly through activation of natural killer cells in a hydrodynamic HBV mouse model (11).
Thus, the potential role of IL-33 may be a context- and disease-dependent manner. Importantly, IL-33 has been demonstrated to modulate CD8+ T cell activity in colorectal carcinoma (36) and nutrient deprivation (8). Thus, we hypothesized that IL-33 could also regulate HBV-specific CD8+ T cell function during chronic HBV infection. To test this possibility, we screened the IL-33 level and ST2 expression in CD8+ T cells in CHB patients, and assessed the effect of IL-33 on viral-specific CD8+ T cell responses in vitro by both phenotypic and functional analysis.
Materials and Methods
Declaration of Helsinki guidelines and approved by the Ethics Committee of The First Hospital of Jilin University. Written informed consent was obtained from each enrolled subject.
Study subjects
A total of 81 treatment-naive CHB patients and 20 healthy controls were enrolled at The First Hospital of Jilin University. All CHB patients were positive for hepatitis B surface antigen (HBsAg) and HBV DNA for more than 6 months. Forty-one CHB patients received entecavir therapy (0.5 mg oral daily). Thirty-two patients achieved virological response (VR) with HBV DNA level <20 IU/mL at 48 weeks posttherapy. Nine patients still had HBV DNA level >20 IU/mL at 48 weeks posttherapy, and were defined as nonvirological response (NVR). Blood samples were collected at baseline, 24, and 48 weeks posttherapy.
The exclusion criteria were infection with hepatitis A virus, hepatitis C virus, hepatitis D virus, hepatitis E virus, and/or human immunodeficiency virus-1. Individuals who were afflicted with autoimmune diseases were also excluded. The baseline clinical characteristics of the subjects are shown in Table 1.
Baseline Clinical Characteristics of Study Subjects
Data are shown as median (range).
ALT, alanine aminotransferase; HBV, hepatitis B virus; HBeAg, hepatits B e antigen.
HBV virological and serological assays
HBV DNA quantification was performed using Cobas® AmpliPrep/cobas® TaqMan® HBV Test, v2.0 (Roche Diagnostics, Rotkreuz, Switzerland) in accordance with the manufacturer's instruction. The detection sensitivity is 20 IU/mL. HBsAg and hepatitis B e antigen (HBeAg) was quantified using ARCHITECT HBsAg and HBeAg QT Reagent Assay Kit (Abbott Laboratories, Abbott Park, IL, USA) in accordance with the manufacturer's instruction.
Isolation of peripheral blood mononuclear cells and purification of CD8+ T cells
All enrolled subjects were bled. Plasma was collected by centrifugation at 1000 rpm for 10 min and was stored at −80°C until use. Peripheral blood mononuclear cells (PBMCs) were isolated using Human Lymphocyte Separation Medium (Ficoll) (Solarbio, Beijing, China) by density gradient centrifugation. CD8+ T cells were purified using Human CD8+ Cell Isolation Kit (Miltenyi, Bergisch Gladbach, Germany) in accordance with the manufacturer's instruction. The purity of sorted CD8+ T cells was more than 95% by flow cytometry analysis.
Cell stimulation and culture
PBMCs or sorted CD8+ T cells were stimulated with recombinant human IL-33 (Peprotech, Rocky Hill, NJ; final concentration: 10, 50, or 100 ng/mL) in the presence of HBV C genotype envelope peptide pool (15 amino acids of each peptide with 5 amino acids overlapping) (final concentration: 2.5 μg/mL) for 96 h.
In coculture experiments, 104 of sorted CD8+ T cells from HLA-A02-restricted CHB patients were cocultured with 5 × 104 of HepG2.2.15 cells in both direct contact and indirect contact conditions as previously described (27). In direct contact condition, CD8+ T cells were directly mixed with HepG2.2.15 cells in the presence of HBV envelope peptide pool, and cultured for 48 h. In indirect contact condition, a Transwell coculture dish (Coring, Corning, NY) was used. CD8+ T cells were added into upper dish with HBV envelope peptide pool stimulation, while HepG2.2.5 cells were seeded into lower dish. The effector and target cells were separated by a 0.4 μm membrane, which allowed the passage of soluble factors only.
Enzyme-linked immunosorbent assay
The expression of IL-33, perforin-1, granzyme B, IFN-γ, and TNF-α was measured using commercial enzyme-linked immunosorbent assay kits (Cusabio, Wuhan, Hubei Province, China) in accordance with the manufacturer's instruction.
Real-time polymerase chain reaction
Total RNA was extracted from sorted CD8+ T cells using TRIzol reagent (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's instruction. First strand cDNA was synthesized with oligo(dT) primer and random hexamers using PrimeScript RT Master Mix (TaKaRa, Beijing, China). Real-time polymerase chain reaction was performed using TB Green Premix Ex Taq II (TaKaRa) in accordance with the manufacturer's instruction. The relative gene expression was semiquantified by 2−ΔΔCt method using Applied Biosystems 7500 System Sequence Detection Software (Applied Biosystems, Foster, CA). The primers for ST2 detection was synthesized as previously described (23).
Cellular proliferation assay
Cellular proliferation was determined by Cell Counting Kit-8 (Beyotime, Wuhan, Hubei Province, China) in accordance with the manufacturer's instruction.
Flow cytometry
PBMCs were stained with anti-CD3-phycoerythrin CF549 (BD Pharmingen, San Jose, CA), anti-CD8-allophycocyanin Cyanine7 (BD Pharmingen), anti-PD-1-fluorescein isothiocyanate (BD Pharmingen), anti-CTLA-4-PE (BD Pharmingen), anti-TIM-3-allophycocyanin (BD Pharmingen), and anti-LAG-3-peridinin-chlorophyll-protein complex Cy5.5 (Invitrogen). The appropriated conjugated IgG antibodies were used as isotype-matched controls. The labeled cells were detected using a Beckman DxFLEX cytometer (Beckman Coulter, Brea, CA), and were analyzed with CytExpert Software (Beckman Coulter) and FlowJo V10 Software (Tree Star, Ashland, OR).
Target cell death
Lactate dehydrogenase (LDH) expression in the supernatants was measured by LDH Cytotoxicity Assay Kit (Beyotime) in accordance with the manufacturer's instruction. The low-level control was defined as LDH expression in HepG2.2.15 cells, while the high-level control was defined as LDH expression in HepG2.2.15 cells with Triton X-100 treatment. The percentage of HepG2.2.15 cell death was calculated using the following formula: (high-level control – LDH expression in the sample)/(high-level control – low-level control) × 100%.
Statistical analysis
Data are expressed as mean ± standard deviation, and they were analyzed by SPSS23.0 Software (SPSS, Chicago, IL). The Student's t test and paired t test was used when two groups were being compared. The one-way analysis of variance and Student-Newman-Keuls-q test were used when more than two groups were being compared. Pearson correlation test was used for correlation analysis. All statistical analyses were based on two-tailed hypothesis tests with a significant level of p < 0.05.
Results
Plasma IL-33 was downregulated in CHB patients
Plasma IL-33 level was first quantified in CHB patients and controls. A robust downregulation of plasma IL-33 was observed in CHB patients compared with controls (Fig. 1A). Given that IL-33 acts by binding to the receptor ST2, we then analyzed the expression of ST2 in CD8+ T cells. There was no significant difference of ST2 messenger RNA in sorted CD8+ T cells between CHB patients and controls (Fig. 1B). IL-33 level was negatively correlated with HBV DNA load (Fig. 1C).

Plasma IL-33 level was downregulated in CHB patients.
The kinetics of plasma level of IL-33 in the longitudinal cohort of entecavir-treated CHB patients was then investigated. There was no significant difference of baseline of plasma IL-33 between VR and NVR (Fig. 1D). Plasma IL-33 expression was significantly elevated 48 weeks posttherapy compared with baseline in VR patients (Fig. 1D). Meanwhile, IL-33 level in the plasma of VR patients was notably increased compared with NVR at 48 weeks posttherapy (Fig. 1D).
IL-33 elevated the abundance of HBV-specific CD8+ T cells
106 of PBMCs from 10 CHB patients were stimulated with HBV envelope peptide pool in the presence or absence of IL-33 (10, 50, or 100 ng/mL) for 96 h. CD8low and CD8high subset within CD3+ T cells was analyzed (Fig. 2A). IL-33 stimulation did not affect PBMC proliferation because there was no remarkable difference of total cell count among groups with different concentrations of IL-33 stimulation and without IL-33 stimulation (Fig. 2B). Addition of IL-33 led to a robust elevation in global CD8+ T cell percentage within CD3+ T cells, which showed a trend of a dose-dependent manner (Fig. 2C). CD8high cell percentage was increased in response to different concentrations of IL-33 stimulation (Fig. 2C). However, only high concentration (100 ng/mL) promoted proliferation of CD8low cells (Fig. 2C).

IL-33 promoted the percentage of HBV-specific CD8+ T cells. PBMCs from CHB patients were stimulated with HBV envelope peptide pool (2.5 μg/mL) in the presence or absence of IL-33 (10, 50, or 100 ng/mL) for 96 h.
IL-33 reduced PD-1 expression in HBV-specific CD8+ T cells
106 of PBMCs from 18 CHB patients were stimulated with HBV envelope peptide pool in the presence or absence of IL-33 (100 ng/mL) for 96 h. The classical immune checkpoint molecules (PD-1, CTLA-4, TIM-3, and LAG-3) expression in HBV-specific CD8+ T cells was investigated. The frequency and intensity of PD-1-expressing HBV-specific CD8+ T cells were notably decreased in the presence of IL-33 (Fig. 3A). Administration of IL-33 did not remarkably reduce the expression of CTLA-4 or TIM-3 in HBV-specific CD8+ T cells (Fig. 3B, C). However, there was little LAG-3 expression in HBV-specific CD8+ T cells in either absence or presence of IL-33 (Fig. 3D).
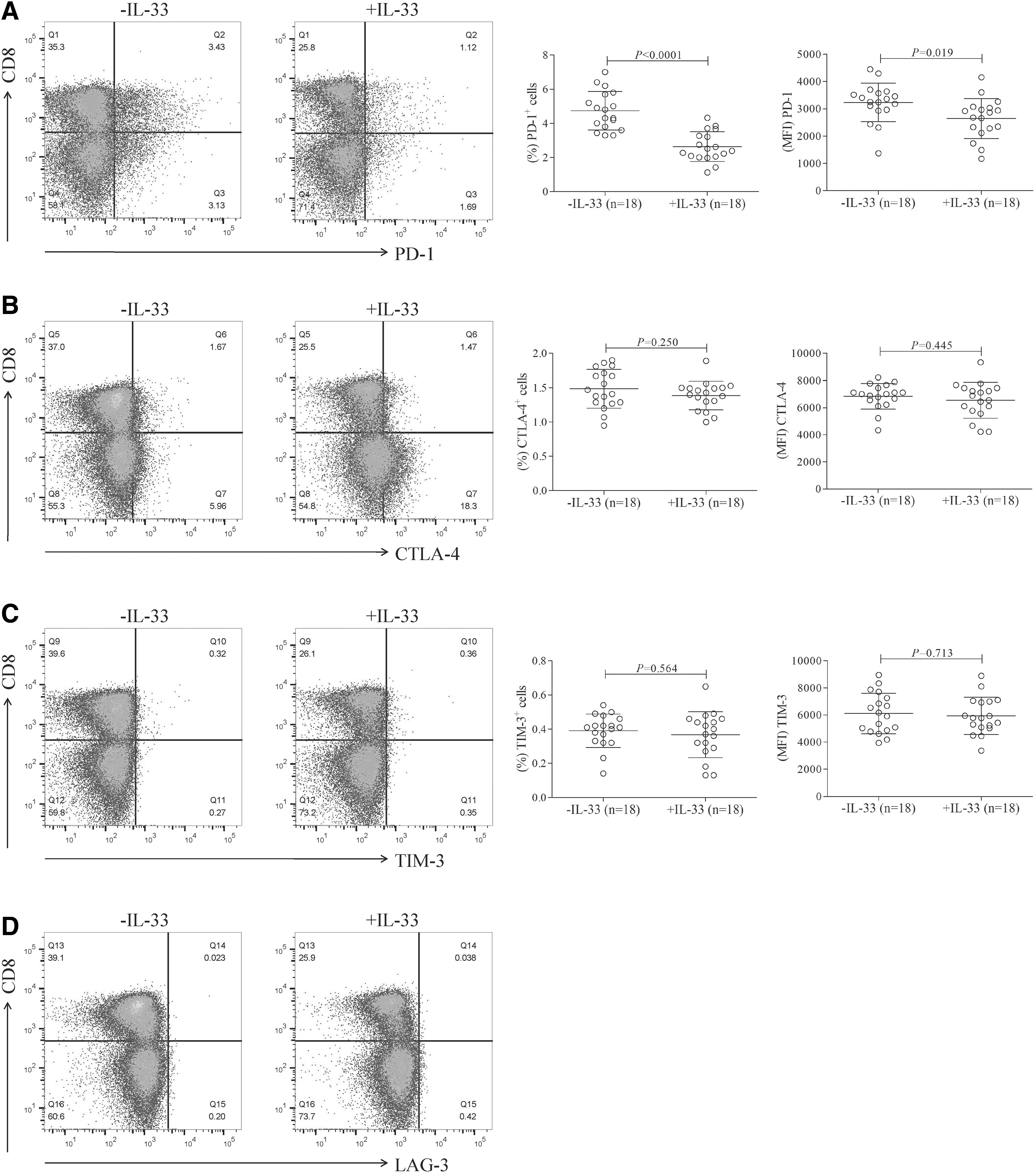
IL-33 reduced PD-1 expression in HBV-specific CD8+ T cells. PBMCs from CHB patients were stimulated with HBV envelope peptide pool (2.5 μg/mL) in the presence or absence of IL-33 (100 ng/mL) for 96 h. PD-1+, CTLA-4+, TIM-3+, and LAG-3+ HBV-specific CD8+ T cells were detected by flow cytometry. The gating strategies for PD-1+, CTLA-4+, TIM-3+, and LAG-3+ cells within CD8+ T cells are shown in
IL-33 enhanced the efficacy of HBV-specific CD8+ T cell-induced cytotoxicity and inhibition of viral replication
104 of sorted CD8+ T cells from 11 HLA-A02-restricted CHB patients, which were stimulated with HBV envelope peptide pool and IL-33 (100 ng/mL) for 96 h, were cocultured with 5 × 104 of HepG2.2.15 cells for another 48 h in both direct contact and indirect contact conditions. The cytotoxicity of CD8+ T cells against HepG2.2.15 target cells was assessed through LDH release assay. In direct contact coculture system, IL-33-primed CD8+ T cells revealed elevated cytotoxicity against HepG2.2.15 cells (Fig. 4A), whereas there was no significant difference in indirect contact coculture system (Fig. 4A). IL-33 administration induced increased amounts of supernatant perforin and granzyme B in direct contact coculture system (Fig. 4B, C).

IL-33 enhanced the cytotoxicity of HBV-specific CD8+ T cells. Sorted CD8+ T cells from 11 HLA-A02-restricted CHB patients were stimulated with HBV envelope peptide pool in the presence or absence of IL-33 (100 ng/mL) for 96 h. CD8+ T cells were washed twice, and 104 of stimulated CD8+ T cells were cocultured with 5 × 104 of HepG2.2.15 cells in both direct contact and indirect contact conditions. Supernatants were harvested 48 h postcoculture.
However, there were no remarkable differences of either perforin or granzyme B secretion between cells with IL-33 and without IL-33 stimulation in indirect contact coculture (Fig. 4B, C). Combination of HBV peptides with IL-33 resulted in robust elevation in abundance of IFN-γ and TNF-α in both direct and indirect contact coculture system (Fig. 4D, E).
In line with this observation, HBV DNA load as well as HBsAg and HBeAg level was significantly decreased in the context of IL-33 administration (Fig. 5A–C).
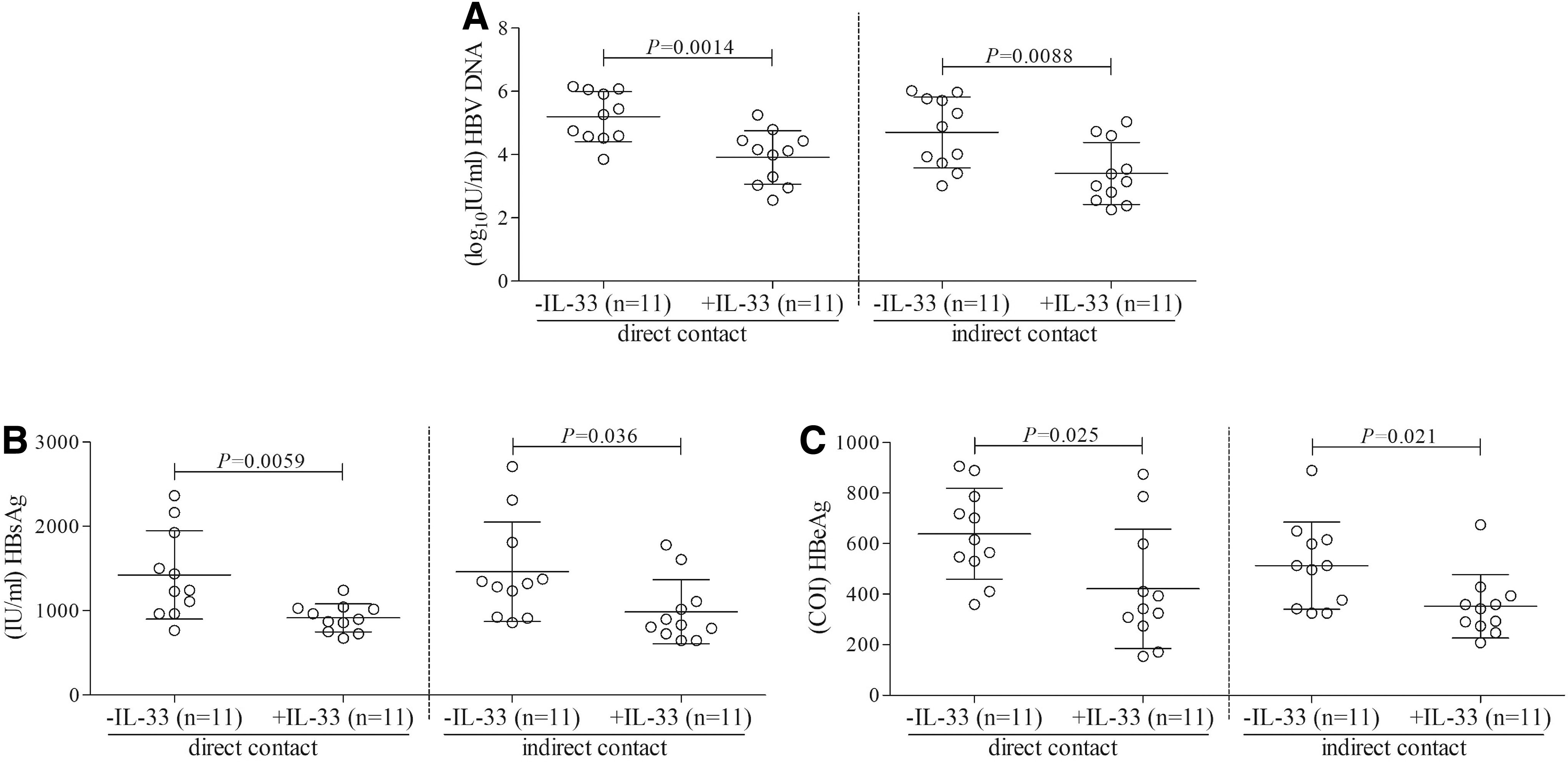
IL-33 promoted the efficacy of HBV-specific CD8+ T cells-mediated inhibition of viral replication. Sorted CD8+ T cells from 11 HLA-A02-restricted CHB patients were stimulated with HBV envelope peptide pool in the presence or absence of IL-33 (100 ng/mL) for 96 h. CD8+ T cells were washed twice, and 104 of stimulated CD8+ T cells were cocultured with 5 × 104 of HepG2.2.15 cells in both direct contact and indirect contact conditions.
Discussion
IL-33 is a well-accepted alarmins, which can either contribute to homeostasis and tissue repair or provoke deleterious uncontrolled inflammation (3). Although IL-33 level was found to be elevated in patients with inflammatory bowel disease and correlated with disease activity index, both pathogenic and protective roles have been demonstrated in experimental colitis animal models (6). Acute viral infection always leads to the upregulation of IL-33. Influenza virus infection induced IL-33 expression in murine lungs (21). Respiratory syncytial virus enhanced IL-33 expression in mouse model of virus-induced exacerbation of inflammation (26). Local activation of IL-33/ST2 axis in nasopharyngeal aspirates and persistent high soluble ST2 concentration was closely associated with severity of viral acute lower respiratory infection in children (28).
Previous studies have shown that serum IL-33 and ST2 varied in different courses of CHB patients, and positively correlated with liver damage (15,34). However, our present report revealed a complete different result in plasma IL-33 expression. We found a reduced IL-33 level in the plasma in CHB patients, while there was no remarkable difference of ST2 expression in CD8+ T cells between CHB patients and controls.
There might be several reasons contributed to the different results. One the one hand, CHB patients enrolled in our study did not suffer with severe liver injury and very limited patients reached 10-fold elevation of serum alanine aminotransferase (ALT). We also did not find significant correlation between IL-33 level and liver inflammation. On the other hand, the results were confirmed by investigating the change of IL-33 during antiviral therapy. CHB patients who achieved sustained VR revealed increased IL-33 level compared with baseline and those who did not achieve VR, indicating that viral replication might contribute to the downregulation of IL-33. This was confirmed by the findings that IL-33 level was negatively correlated with HBV DNA. Taken together, decreased IL-33 level was associated with viral replication in CHB patients.
Viral specific CD8+ T cells are not only capable to control HBV infection but also to eliminate HBV-infected cells (14). However, HBV-specific CD8+ T cells were dysfunctional or exhausted during persistent HBV infection. Due to the findings in the correlation between IL-33 and viral replication, the regulatory function of exogenous IL-33 to HBV-specific CD8+ T cells from CHB patients was then assessed in vitro. IL-33 suppressed fibroblastic cell line growth in confluent cells, however, promoted cell growth after the start of cell proliferation (32). Importantly, IL-21 promoted HBV-specific CD8+ T cells in a dose-dependent manner (31).
Thus, we analyzed PBMCs proliferation in response to different concentrations of recombinant IL-33 stimulation. Exogenous IL-33 stimulation did not promote PBMCs proliferation. However, IL-33 enhanced the percentage of HBV-specific CD8+ T cells in different concentrations. Only higher concentration of IL-33 increased both CD8high and CD8low subset. CD8high cells were highly cytolytic via expressing high level of perforin and granzyme, while CD8low cells were poorly cytolytic and expressed low levels of perforin and granzyme B and C (17,18). Both CD8high and CD8low cells secreted IFN-γ and IL-4 (17,18). It might be that even lower concentration of IL-33 also promoted cytolytic function of CD8+ T cells. Thus, 100 ng/mL of IL-33 was then used for the following experiments.
Exposure to IL-33 augmented the percentage of HBV-specific CD8+ T cells in PBMCs from CHB patients. Furthermore, immune checkpoint inhibitor could restore exhausted HBV-specific CD8+ T cells via different mechanisms (2,7,35). We found that IL-33 stimulation led to downregulation of inhibitory receptor PD-1 in HBV-specific CD8+ T cells, implying that IL-33 may help rescue dysfunctional/exhausted HBV-specific CD8+ T cell activity during persistent HBV infection. This observation was similar to the previous findings that IL-21 potently restored the capacity of HBV-specific CD8+ T cell by PD-1 and TIM-3 downregulation (31). It was reported that transcriptional factor T-bet could induce IL-33 receptor expression directly, leading to the direct promotion of antiviral Th1 responses (1). T-bet deficiency facilitated the elevated PD-1 expression in exhausted CD8+ T cells in chronic viral infection (16). Thus, we speculated that IL-33 might mediate T-bet expression and subsequently reduced PD-1 expression to overcome the dysfunction of HBV-specific CD8+ T cells.
Overall, IL-33 might restore the exhausted HBV-specific CD8+ T cells by inhibition of PD-1 expression and improvement of expansion capacity during chronic HBV infection.
Both cytolytic and noncytolytic mechanisms contribute to HBV clearance (27). On the one hand, polyclonal HBV-specific CD8+ T cells resolved HBV infection and developed ALT flares during acute HBV infection, suggesting that the cytolytic mechanism are involved in inhibition of viral replication (30,37). This process was required direct cell-to-cell contact, and perforin-granzyme signaling pathway contributed to destroy HBV-infected hepatocytes (22,33). On the other hand, CD8+ T cells could also secrete cytokines to mediate noncytolytic control of HBV replication without destructing the infected cells (13). Phillips et al. used in vitro coculture model to independently assess the cytolytic and noncytolytic functions of HBV-specific CD8+ T cells and to dissect the roles of these two functions in controlling of HBV replication in target cells (27).
By using these coculture systems, we tried to assess the regulatory function of IL-33 to cytolytic and noncytolytic activities of HBV-specific CD8+ T cells. We found that administration of IL-33 strongly suppressed HBV replication and release of HBV antigens in both systems, indicating that IL-33 contributed to both functional regulations of viral-specific CD8+ T cells during chronic HBV infection. However, IL-33 did not induce elevated percentage of target cell death, indicating that increased cytokine expression might be insufficient to destroy infected hepatocytes, but could induce the inhibition of viral replication. These findings suggested that IL-33 may contribute to control HBV by both facilitating the cytolytic effector function and secretion of cytokines by HBV-specific CD8+ T cells.
In conclusion, the current data indicated that chronic HBV infection induced insufficient secretion of IL-33. Exogenous IL-33 reinvigorated antiviral activity of HBV-specific CD8+ T cells by enhancement of expansion capacity and cytokine production as well as by inhibition of PD-1 expression. The comprehensive understanding of IL-33 regulation to CD8+ T cells may facilitate the development of immunotherapeutic strategies to chronic HBV infection.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received.