
Abstract
The immune system of active and inactive chronic hepatitis B, as prolonged forms of hepatitis B, is unable to eradicate hepatitis B virus (HBV) from the infected hepatocytes completely. Toll-like receptors (TLRs) play key roles in the viral recognition and promotion of appropriate immune responses. The molecules also participate in the alteration of the target cell functions and transformation. TLR2 is the unique molecule that makes either homodimer or heterodimer with TLR1 and 6 and shows variable roles against viral infections. Therefore, it has been hypothesized that TLR2 may participate in both immune response against HBV and induction of the virus-related hepatic complications. The studies confirm the hypothesis and revealed that TLR2 is not only one of the main molecules altering the course of HBV infection, but also plays key roles in induction of hepatocellular carcinoma (HCC) and liver cirrhosis. However, recent studies demonstrated that the molecule can fight against HCC and liver cirrhosis. Collectively, it appears that nutrition habits, TLR2 gene polymorphisms, gut microbiome, HBV antigens, and activation of other receptors may play key roles in the determination of TLR2 functions.
Background
Hepatitis B is a prevalent human liver infectious agent that is induced by a hepatotropic virus entitled hepatitis B virus (HBV) (97). HBV, as a member of the Hepadnaviridae family, contains a small DNA virus in the core protein as its genome and replicates through an RNA intermediate, and can integrate its genome in the host genome (43). HBV contains some proteins, including hepatitis B surface antigen (HBsAg), hepatitis B e antigen (HBeAg), and hepatitis B core antigen (HBcAg) (43). The C gene encodes both HBcAg and HBeAg.
The virus induces several clinical forms, including acute, fulminant, chronic, and occult hepatitis B (43). In the chronic and occult hepatitis B, as prolonged forms, the immune responses are unable to clear the virus from both hepatocytes and sera (97). The prolonged term HBV infections alter cell conditions to be transformed as hepatocellular carcinoma (HCC) or inactivate the cell functions by induction of fibrosis in the liver tissue that is named liver cirrhosis (27). It has been demonstrated that HBV induces the alteration in the cell functions leading to HCC and liver cirrhosis (27).
Moreover, the virus produces some molecules to alter immune responses, which can be associated with various phenomena, including immune evasion and also induction of HCC and liver cirrhosis (82). However, it appears that the host immune responses to the virus are the main variables to determine the outcome of the infection. For example, an appropriate immune response can be associated with eradication of HBV and clearance of the hepatocytes (82). Although prolonged nonprotective immune responses are the main causes of persistent HBV infections and alterations in hepatocyte, intracellular signaling is the main cause of transformation of the cells (1,4,5).
Innate immunity produces the first antiviral barriers against microbial infections and activates adaptive immunity to clear the infections (8,29,62). Innate immune cells recognize some antigens of the microbes that are named as pathogen-associated molecular patterns (PAMPs) through their pathogen recognition receptors (PRRs) (8,61). The receptors are located on the cytoplasmic membrane, in the intracellular organelles, such as vesicles, and in the cytoplasm (73). Recent studies revealed that these receptors can be important in determining the outcome of infectious diseases (73). Among the PRRs, toll-like receptors (TLRs) are more commonly evaluated regarding their roles in the viral infection outcomes.
TLRs express on both cytoplasmic membrane and in the membrane organelles such as the endosome, the endoplasmic reticulum and the lysosome, and recognize the PAMPs in the vesicles and outside of the cytoplasm to induce appropriate immune responses (32). Therefore, the molecules are the important members of the immune puzzle to HBV.
In addition, as mentioned previously, HCC and liver cirrhosis can be induced as a result of the virus functions and immune responses. Because TLRs are the key molecules that participate in the immune responses to HBV, it has been hypothesized that the molecules also play key roles in either HBV eradication or induction of the virus complications, including HCC and liver cirrhosis. We have discussed the roles of TLRs in the outcome of HBV infection previously (6,28,67,98). However, several recent studies investigated the roles played by TLR2 in both anti-hepatitis B immune responses and pathogenicity of the immune responses during hepatitis. This review article summarized the updated information on the roles of TLR2 in the pathogenicity of hepatitis B.
Toll-Like Receptors
Ten TLRs are found in human, and the functions of the molecules have been well described (20 –22,35). TLRs are expressed as dimer on the cell membrane and intracellular vesicles (36). TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, and TLR9 produce homodimer, whereas TLR1 and TLR6 produce heterodimer with TLR2. The dimmers recognize various ligands by their extracellular domains. For example, TLR2/2 recognizes lipoteichoic acid, bacterial peptidoglycan, lipoprotein, porins, viral glycoproteins, and hemagglutinin (36), and TLR1/2 and TLR2/6 heterodimers can trigger bacterial tri- and diacylated lipopeptides, lipopeptides, and lipoteichoic acid (2,13).
TLR2 also can detect Pam3Cys, as an important ligand for induction of TLR2-related intracellular signaling pathways. The main ligands for TLR3/3, TLR4/4, TLR5/5, and TLR9/9 are double-strand RNA, lipopolysaccharide, bacterial flagellin, and CpG oligonucleotides, respectively. TLR7/7 and TLR8/8 are unique receptors that detect single-strand RNA, especially viral RNA (76). However, the TLRs are not limited to these ligands and in some cases, their recognitions have overlaps. TLR1/2, TLR2/2, TLR2/6, TLR4/4, and TLR5/5 are expressed on the cytoplasmic membrane, whereas TLR3/3, TLR7/7, TLR8/8, and TLR9/9 recognize the intracellular vesicle ligands (66).
TLRs use two main intracellular signaling pathways that are named as their adaptor proteins, toll/interleukin-1 (IL-1) receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF), and myeloid differentiation primary response (MYD88) (66). TLR1/2, TLR2/2, TLR2/6, TLR5/5, TLR7/7, TLR8/8, and TLR8/8 usually use MYD88, TLR3/3 uses TRIF and TLR4/4 uses both TRIF and MYD88 dependent pathways. The details of the signaling pathways have been reported in our previous review articles (6,28,67,68,98). In brief, TLR2 induces activation of interleukin-1 receptor associated kinase-4 (IRAK4), IRAK1, and IRAK2 through MYD88 adaptor protein.
The activation of the complex leads to phosphorylation of tumor necrosis factor (TNF) receptor-associated factor-6 (TRAF6), which is the activator of transforming growth factor b-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), and TAB2 complex. The recent complex is the activator of several molecules and pathways, including nuclear factor kappa B (NF-κB), extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK), and MAPK, which play important roles in the expression of TLR2 target genes [34, 35]. Figure 1 illustrates the signaling pathway of TLR2.
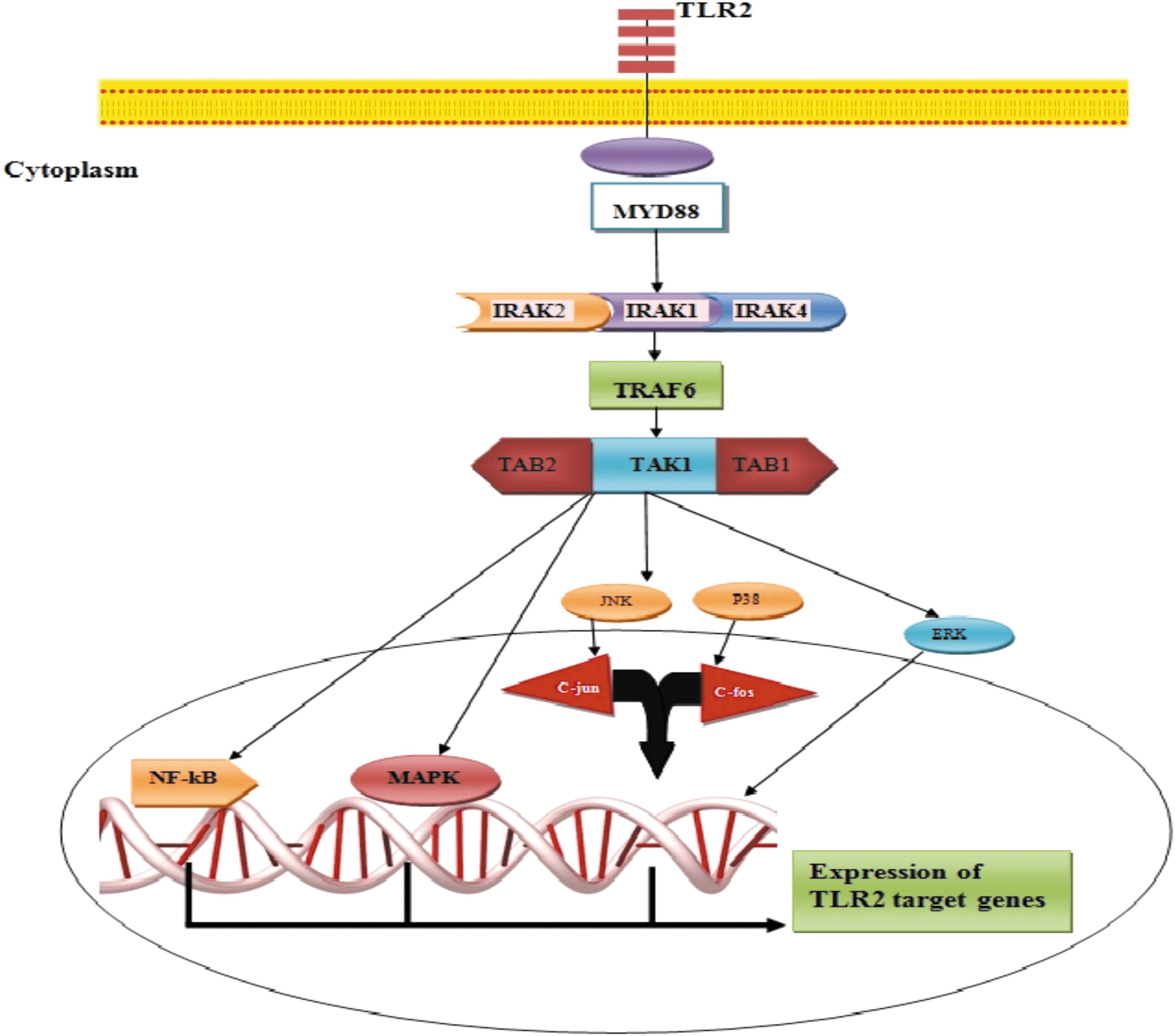
TLR2 intracellular signaling pathway. TLR2 induces activation of IRAK4, IRAK1, and IRAK2 through MYD88 adaptor protein and then TRAF6. Activation of TRAF6 leads to phosphorylation of TAK1, TAB1, and TAB2 complex, which is the activator of several molecules and pathways, including NF-κB, ERK, JNK, p38 MAPK, and MAPK, and finally expression of TLR2 target genes. ERK, extracellular signal-regulated kinase; IRAK4, interleukin-1 receptor-associated kinase-4; JNK, c-Jun N-terminal kinase; TAB1, TAK1-binding protein 1; TAK1, transforming growth factor b-activated kinase 1; TLR, toll-like receptor; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; TRAF6, tumor necrosis factor receptor-associated factor-6. Color images are available online.
TLR Dimmers 1/2, 2/2, and 2/6 Participate in the Defense Against HBV
TLRs' main roles are to alter immune cell and hepatocyte functions to overcome the infections. It has been reported that TLRs significantly participate in the recognition of HBV-related molecules and induce innate and then adaptive immune responses (52).
HBcAg is a HBV glycoprotein in the center of HBV particle that can be recognized by TLR2 to induce pro-inflammatory reactions (13). Accordingly, G1896A pre-core mutations were associated with increased expression of TLR2 (60). An animal model study also demonstrated that TLR2 is an important PRR to induce innate immunity against hepadnaviruses (100). Using TLR2 ligands as adjuvant in the vaccination against HBV revealed that the ligands increased both cellular and humoral immunity against HBV, significantly (14). Another study revealed that TLR2-deficient mice were unable to kill HBV by T cytotoxic cells (55).
Therefore, it appears that TLR2 contributes significantly to the activation of either innate or adaptive immunity against HBV. Accordingly, the in vitro studies approved the hypothesis. For example, a study on the PLC/PRF/5 and Huh1 cell lines showed that TLR2 elevation is associated with decreased expression of HBsAg (17). Thus, it seems that TLR2 can induce some signaling pathways that lead to inactivation of HBV-related translation and it may happen directly through TLR2-related signaling pathways and indirectly through upregulation of type-1 interferons. Consequently, it was proved that TLR2 is expressed by hepatocytes (64).
Expression of TLR2 can be associated with HBV copy number in the hepatocytes and under treatment with synthetic isosteviol-derived analogues against the HBV will be decreased (23). Thompson et al., reported that TLR2 engagement on the hepatoma cell lines led to inhibiting HBV replication (75). A study by Zhang et al. showed that treatment of primary human hepatocytes with both Pam3Cys, a synthetic ligand for TLR2, and HBV particles led to phosphorylation of ERK, JNK, and p38 MAPKs, which result in activation of transcription factors. Interestingly, they revealed that, HBV particle-induced pathways could be neutralized using the TLR2-specific antibodies (101). In addition, it has been demonstrated that in the chronic hepatitis B (CHB) patients with resistance to entecavir, the pegylated interferon (PEG-IFN)-α2a plays a positive role in the fight against HBV (92). Studies revealed that PEG-IFN-α2a plays this role through several mechanisms, including TLR2-dependent pathways (92).
In addition to activation of innate immunity in the hepatocytes, some researchers reported that TLR2 significantly increased functions of immune cells against HBV. For instance, an in vivo study revealed that HBV particles activate B cells through the TLR2-MYD88 axis (38). In addition to humoral immunity, preactivation of TLR2 can be associated with CD8+ T Cell responses and promotes HBV clearance in the animal models (47). A study by Lucifora et al. revealed that Pam3CSK4 (TLR1/2-ligand) can activate hepatocytes and immune cells to produce antiviral cytokines (53). Based on the results, TLR2 appears to be a key molecule on both hepatocytes and immune cells that participates in the recognition of HBV antigens and activation of either innate or adaptive immunity against HBV.
The potential roles played by TLR2 were also confirmed by the fact that the polymorphisms within TLR2 gene are significantly associated with immune responses and outcome of HBV infection. For example, a study revealed that rs3804099 and rs4696480 TLR2 genotypes were significantly associated with altered production of TNF-1 and interleukin (IL)-6 by innate immune cells (46). A research on a Chinese population proved that the rs3804100 polymorphism in the TLR2 gene is associated with increased humoral immunity to the HBV antigens (86). Another study revealed that TLR2 rs3804100 polymorphism acts as a protective factor for HBV-related HCC (9).
Owing to the important roles of TLRs against HBV, in the cases of prolonged forms, the virus can escape recognition by TLRs or suppress their internal signaling. For example, previous studies reported that HBV infection led to downregulation of either mRNA or protein levels of TLR2 in the CHB patients (11,65). Increased HBV copy numbers were also associated with decreased expression of TLR2 in the CHB patients (95). Our previous study demonstrated that IRAK4 mRNA levels, a TLR intracellular molecule, were also decreased in the peripheral blood mononuclear cells (PBMCs) of CHB patients (59). TLR2 also participates in inhibition of a pathological storm form of immune responses against HBV, especially in fulminant forms (18). It appears that the pro-inflammatory responses in the CHB patients may be related to the TLR2.
Moreover, researches showed that patients with chronic hepatitis B express more PBMCs and hepatocyte levels of TLR2 (16,87). Although elevated levels of TLR2 were demonstrated in the CHB patients, its molecular signaling may be defected or suppressed by HBV, which leads to defected immunity against HBV. For example, a study by Heiberg et al. showed the hepatocytes from CHB patients produced significantly lower IFN-α in comparison with healthy controls, when they were stimulated with TLR2 ligands (19). It appears that HBV and its related molecules directly target the TLR2 and its related signaling molecule to overcome the immune response.
Hence, HBsAg can suppress TLR2-induced IL-12 production through blocking the JNK-MAPK pathway (84). HBeAg also suppresses the signaling pathways by disrupting the homotypic TIR/TIR interactions, which leads to activation of NF-κB and production of IFN-β (34). A study by Wang et al. revealed that HBeAg suppresses NF-κB through interrupting K63-Linked ubiquitination of NF-κB essential modulator (NEMO) (85). In addition to disruption of intracellular signaling, HBeAg can downregulate TLR2 expression on the CHB patient peripheral monocytes, hepatocytes, and liver macrophages, whereas it was increased in the HBeAg negative patients when compared with healthy controls (79).
Akbal et al. also confirmed the roles played by HBeAg on the TLR2 levels and reported that serum levels of the molecules are higher in the HBeAg negative group, but not positive, compared with controls (3). HBeAg also inhibits hepatocytes IL-1β mediated NF-κB activation (88). HBV-related antigens can induce cellular immunity against the virus in an animal model (55), maybe through inhibition of the MyD88-IRAK4 axis, the main intracellular signaling pathway of TLR2 (77).
HBV is also able to reduce activity and expression of TLR2 and its related molecules indirectly. For instance, HBV alters monocyte functions to produce IL-10 (69) to downregulate TLR2 and its related molecules through the JAK1/STAT3 signaling pathway (49). According to the results, it may be hypothesized that TLR2 can be considered as a main PRR, which participates in response against HBV.
However, this conclusion needs more consideration. Because in contrast to the mentioned studies, some reports demonstrated that HBV can trigger TLR2 to increase expression of anti-inflammatory cytokines, such as IL-10. Li et al. reported that activation of Kupffer cells by TLR2/HBcAg interaction led to immune tolerance to HBV-related antigens through increased IL-10 production and then exhaustion of anti-HBV CD8+ T cell (37). Another research showed that expression of TLR2 was increased on the B-regulatory lymphocytes, the cells participate in suppression of immune responses against HBV in the CHB patients (81). It has been reported that intrahepatic myeloid-derived cells (iMDCs) participate in the promotion of cellular immunity against HBV (30). Palmitoyl-3-cysteine-serine-lysine-4 (P3C), as TLR2 ligand, suppresses the function of iMDC and induces its tolergenic properties (48).
According to the results, it seems that the roles of TLR2 in the HBV infection are controversial. For example, Isogawa et al. demonstrated that ligands for several TLRs with the exception of TLR2 were capable of inhibiting HBV infection (24). Based on the previous studies, it appears that some factors may determine the outcome. A study by Janovec et al. revealed that activating TLRs using dual-acting agonists is more effective than one agonist to induce PBMC-produced cytokines, and then inhibit HBV production in primary human hepatocytes (26).
In addition, specific dietary fibers can be considered as a variable to alter responses of TLR2. For example, a study showed that specific dietary fibers significantly participate in the TLR2 functions during infection with HBV (80). Another reason for various functions of TLR2 during hepatitis B is related to two important factors: HBV genotype and pretreatment with immune inducers (78). In addition, as mentioned previously, genetic variations in the TLR2 and its related molecules can affect their functions. Therefore, it seems that both genetic and environmental factors have an effect on the TLR2 properties against HBV.
TLR2 Roles in the Hepatitis B-Associated Hepatic Complications
HCC and cirrhosis of the liver are the main complications of CHB (33). In addition, it has been demonstrated that TLR2 is a major molecule that participates in the induction of inflammation during an acute form of hepatitis B, which can lead to liver cirrhosis and HCC (7). In addition, it has been documented that TLR2 signaling is associated with hyperlipidemia (103), which is a main risk factor for induction of liver complications, such as HCC (63). Therefore, this can be considered as a symptom for the participation of TLR2 in the pathogenesis of liver cirrhosis and HCC, through induction of inflammation and hyperlipidemia.
Cheng et al. reported that HBsAg promotes the invasion of HBV-related HCC through increased expression of TLR2 (12). TLR2 expression was also associated with promotion of some oncogene expressions in the HBsAg-transgenic mice (Alb/HBs) (99). However, the roles played by TLR2 in the HCC have not been confirmed. Accordingly, recent studies reported that TLR2 could inhibit HCC induction. Luo et al. reported that TLR2 directly mediates activation of the Hippo signaling pathway in the hepatocytes (54). It has been documented that Hippo signaling pathway inhibits development of HCC (51).
TLR2 may also participate in the liver cirrhosis induction in the CHB patients. Guo et al. showed that high grade of necroinflammatory activity was associated with the intensity of TLR2 expression on the HBV-infected hepatocytes (16). Increased expression of TLR2 was also associated with CHB-related liver failure (83). Elevated TLR2 expression on the hepatocytes of the patients suffering from cirrhosis was also reported by Soares et al. (72). Moderate and severe grades of chronic hepatitis B are associated with increased risks of liver cirrhosis and interestingly their TLR2 expression was increased when compared with mild disease (96).
Similar to HCC, there are some controversial results regarding the roles played by TLR2 in the liver cirrhosis. It has been reported that M2 macrophages are the main cells that participate in the pathogenicity of HCC and liver cirrhosis (89). Yi et al. reported that HBcAg increased the expression of pro-inflammatory cytokines by M2 macrophages and decreased expression of M2-related molecules through the TLR2 pathway (94). Lian et al. also revealed that TLR2 expression was not different in the patients with liver cirrhosis in comparison to the CHB patients (41).
Thus, it seems that TLR2 and its related molecules may show various properties during prolonged forms of hepatitis B infection. We have discussed the plausible mechanisms used by TLR2 to induce HCC and liver cirrhosis previously (6).
Here we summarize and update them: The MYD88-dependent intracellular signaling can be a main inducer of HCC through NF-κB and JNK activation (42,56). HBV elevates expression of cholesterol metabolism-related genes in TLR2 pathway-dependent manner (40), cholesterol and its metabolic pathway are the main risk factors for the liver cirrhosis (25). TLR2 may be involved in the induction of cirrhosis and HCC through upregulation of pro-inflammatory cells and molecules (57,74). Accordingly, TLR2 participates in the pathogenicity of hepatitis B through increased differentiation of Th17, which is associated with disease aggravation of liver cirrhosis (90). The results were confirmed by Zhao et al., and revealed that TLR2 significantly directed differentiation of Th cells to Th17 (102).
However, due to the fact that the studies regarding the roles played by TLR2 in the liver complications in the HBV-infected patients are limited to the mentioned studies, there are several studies regarding the roles of the molecule and its related pathways in the induction of HCC and liver cirrhosis in the non-HBV-infected patients (50,58,70,71,72,91,93).
Interestingly, the roles played by TLR2 in the non-HBV-infected patients with HCC and liver cirrhosis were also controversial. For example, Li et al. reported that TLR2 limits development of HCC through modulation of IL18-mediated immunosuppression (39). Another study by Chen et al., in an in vitro study, revealed that TLR2 signaling pathway could inhibit the proliferation of HCC cells (10). Lack of TLR2 can also be associated with increased risk of HCC in the animal models (45). Lack of TLR2 may be associated with attenuation of autophagic flux and leads to the development and progression of HCC (44).
The main mechanisms for determination of the TLR2 roles in the outcome of HCC and liver cirrhosis are yet to be clarified. However, authors present some plausible mechanisms here: Nutrition habits may be a main reason for various roles of TLR2 during HCC and liver cirrhosis. For instance, French et al. demonstrated that alcohol and nutrition significantly affect the roles played by TLR2 during HCC (15). TLR2 polymorphisms may affect the functions of the molecule in the hepatocytes and immune cells. As mentioned previously, TLR2 polymorphisms were significantly associated with outcome of HBV infection. It has been reported that some CHB patients express HBsAg and HBeAg, and the molecules can affect TLR2 functions. It has been reported that gut microbiome may affect TLR2 functions. For example, Lactobacillus acidophilus can suppress TLR2/STAT-3/P38-MAPK pathway in an animal model (31). The intracellular signaling of TLR2 is overlapped by other intracellular signaling and, hence, can be modulated by pathways. In addition, the effects of other cell signaling on the protein expressions can affect TLR2 functions. Accordingly, a study showed that stimulation of the IL-1 receptor and TLR 2 simultaneously, suppresses HBV replication in hepatoma cell lines (75). Thus, activation of the hepatocytes by other molecules may affect TLR2 functions.
The plausible roles played by the variables on the outcome of TLR2 functions are presented in the Table 1. It appears that both genetic and environmental factors can affect the outcome of TLR2 functions and its effects on the hepatocyte life.
The Plausible Mechanisms Involved in the Determination of the Toll-Like Receptor 2 Roles in the Outcome of Hepatocellular Carcinoma and Liver Cirrhosis
The data demonstrated that several factors may affect the roles played by TLR2 in the patients with chronic hepatitis B.
HBeAg, hepatitis B e antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; IL-10, interleukin-10; JNK-MAPK, c-Jun N-terminal kinase-mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; TIR, toll/interleukin-1 (IL-1) receptor.
Conclusion and Future Prospects
Our previous review article revealed that TLR2 can induce immune response against HBV and also is an inducer of HCC and hepatic cirrhosis. However, the recent article showing that TLR2 can be a protective molecule against HCC and hepatic cirrhosis. Both genetic and environmental factors can affect the outcome of TLR2 functions. Thus, it may be hypothesized that future immunotherapies could focus on regulation of the factors to direct TLR2 functions against HBV and its related complications. In contrast with our previous review article, the updated information suggests that TLR2 and its signaling molecules act as both inducers and inhibitors of HCC and liver cirrhosis. However, the results are from non-HBV-infected HCC patients, hence, more studies in the HBV-related HCC are needed to clarify the controversial reports.
Authors' Confirmation Statement
Dr. M.N. and Dr. A.K. are from Islamic Azad University, Kerman Branch (Kerman, Iran), and Dr. M.K.A. is from the Rafsanjan University of Medical Sciences (Rafsanjan, Iran), where education and research are the primary functions.
Footnotes
Authors' Contributions
M.N. collected related articles. A.K. and M.K.A. wrote and completed the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The Islamic Azad University, Kerman Branch and Rafsanjan University of Medical Sciences funded grants to this project.